
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical initiation of electron-catalyzed phenanthridine synthesis by trifluoromethylation of isonitriles†
M.
Lübbesmeyer
,
D.
Leifert
,
H.
Schäfer
and
A.
Studer
 *
*
Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: studer@uni-muenster.de
First published on 12th February 2018
Abstract
The electron-catalyzed formation of phenanthridines starting from isonitriles initiated by an electrochemical reduction of the Togni reagent is presented. The required number of faradays per mole of starting material and the respective yields clearly show the catalytic character of the electron in this reaction. The mechanism is supported by cyclic voltammetry experiments.
Several radical reactions can be described by catalytic cycles in which a single electron formally acts as the catalyst.1 Examples developed in our group include perfluoroalkylation and aroylation reactions comprising a base promoted homolytic aromatic substitution (BHAS),2,3 hydrodehalogenations,4 cross dehydrogenative couplings via BHAS5 and cascade reactions.6 The concept was adapted by other groups, e.g. for the description of a benzothiazine formation by a sulfur insertion reaction with enaminones7 and perfluoroalkylations of organic sulfides.8 A tandem carbodifluoroalkylation/radical cyclization developed by Zhu et al. where difluoroacyl radicals add to methylene-2-oxazolines represents another nice example along these lines.9
The goal of the current study was to confirm the catalytic role of the electron by electrochemical initiation of a recently developed radical trifluoromethylation reaction. Comparison of the electric charge quantity with the respective reaction yield is a measure for the turn over number of the electron acting as a catalyst. In a similar way, electron catalyzed radical-nucleophilic aromatic substitution reactions (SRN1) have been previously investigated.10 Further examples for which the catalytic character of the electron could be demonstrated include E/Z-isomerization reactions,11 oxidations of primary and secondary alcoholates into aldehydes and ketones12 and cyclodimerizations between aryl vinyl sulfones as well as unsaturated ketones.13
As a test reaction we chose the radical trifluoromethylation of biaryl isonitrile 1a with the Togni reagent 2 comprising an intramolecular BHAS to provide phenanthridine 3a (Fig. 1).2 The phenanthridine structural motif plays a key role in several natural products.14 Its importance is underlined by numerous studies related to its synthesis.15
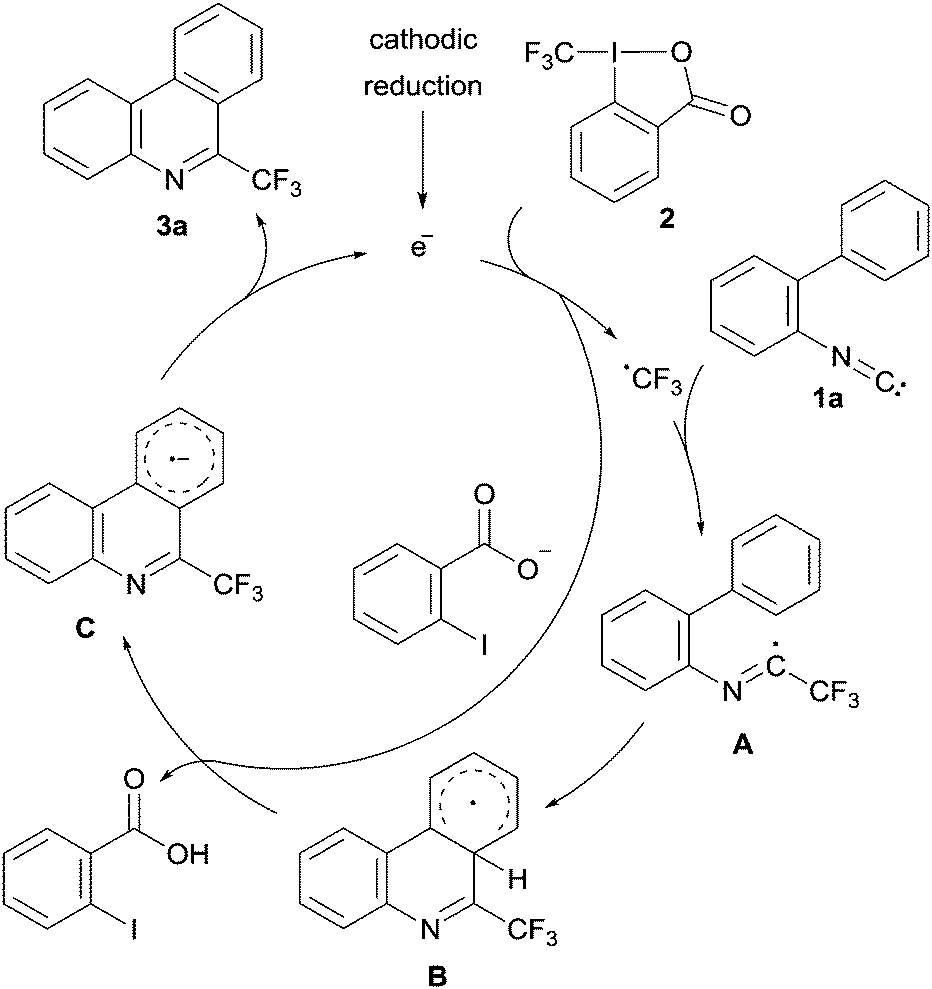 | ||
| Fig. 1 Suggested mechanism for the formation of 6-(trifluoromethyl)-phenanthridine from 2-isocyano-1,1′-biphenyl. | ||
For the investigated reaction we previously suggested a mechanism in which the electron formally acts as a catalyst (see Fig. 1). A trifluoromethyl radical, generated upon single electron reduction of 2, first adds to the isonitrile functionality of the 2-isocyano-1,1′-biphenyl 1a leading to the imidoyl radical A. After cyclization, the resulting cyclohexadienyl radical B is deprotonated by ortho-iodobenzoate which is formed as a byproduct in the initial SET reduction of the Togni reagent. The radical's pKa value was calculated to be significantly lower relative to the pKa value of the ortho-iodobenzoic acid.2a The resulting radical anion C is a reducing agent that can sustain the chain by an intermolecular electron transfer to the Togni reagent yielding the targeted 6-(trifluoromethyl)phenanthridine 3a. This mechanism was additionally supported by trapping experiments of the CF3-radical with TEMPO.2a Moreover, in a recent study in collaboration with Gorbanev, O’Connel and Chechik we could show that the described reaction can be efficiently initiated by a helium plasma indicating the possibility of an electrochemical initiation.16 However, the measurement of an electric current and, therefore, confirmation of the electron's catalytic character remained unexplored.
Our studies commenced by analyzing both substrates 1a, 2 and the product 3a by cyclic voltammetry. The Togni reagent 2 is irreversibly reduced at a peak potential of −1.58 V against the ferrocene/ferrocenium redox couple (Fig. 2, left). In the reverse scan an irreversible oxidation at 0.91 V vs. Fc/Fc+ is observed. We ascribe this to the formation of ortho-iodobenzoate, which is irreversibly oxidized (Fig. 2, right). To support this proposal, a solution of tetrabutylammonium ortho-iodobenzoate was freshly prepared by deprotonation of the corresponding acid with tetrabutylammonium hydroxide. Indeed, CV analysis revealed a similar oxidation peak at 0.96 V vs. Fc/Fc+.
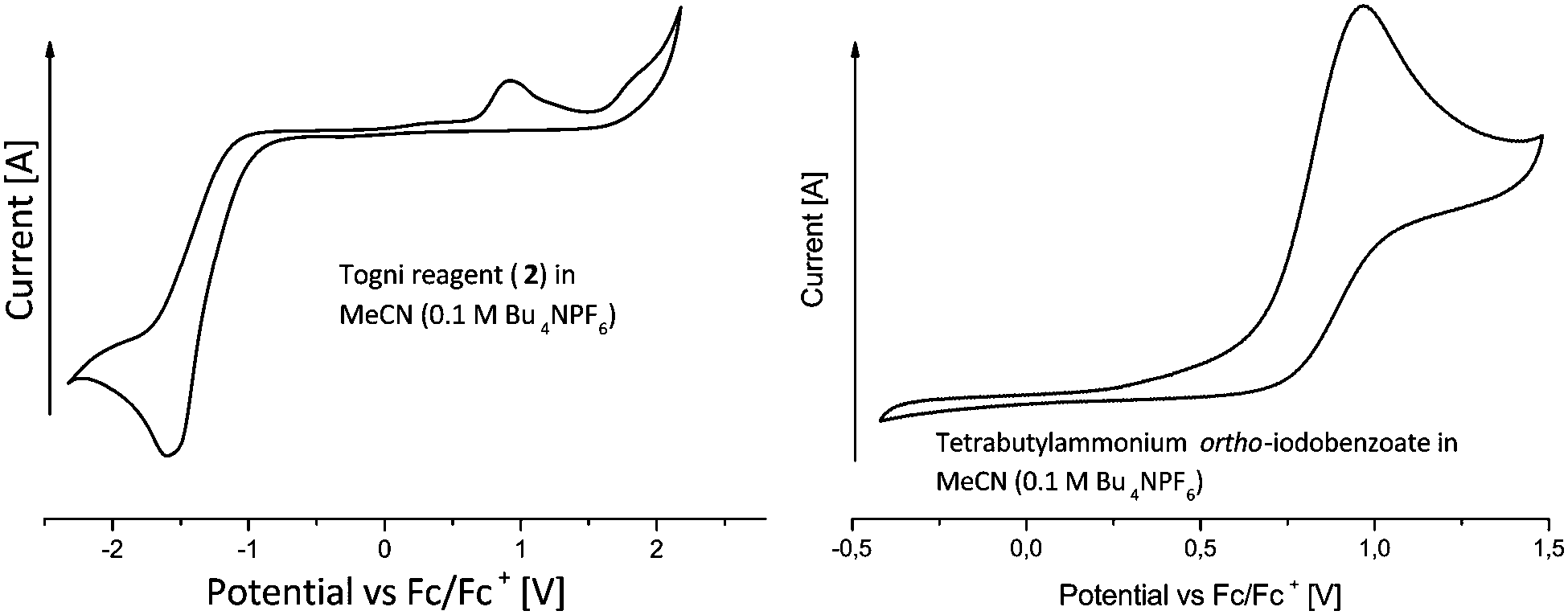 | ||
| Fig. 2 Cyclic voltammetry of Togni reagent 2 (left) and ortho-iodobenzoate (right). | ||
To examine the dioxane's electrochemical window, dioxane was added to an acetonitrile solution containing Bu4NPF6 (0.1 molar, see the ESI†). The CV measurement revealed that dioxane is oxidized at less positive potentials as compared to acetonitrile (inset approx. 1.6 V vs. Fc/Fc+). Substrate 1a and product 3a are irreversibly oxidized at peak potentials of 1.78 and 1.89 V, respectively. To promote solvent oxidation and prevent substrate or product oxidation a platinum wire anode was utilized. This way the undivided cell is changed to a quasi-divided cell and the substrate and product are protected against oxidation. Since only catalytic amounts of electric charge have been used, we regard possible losses in yield due to substrate or product oxidation to be low.
The phenanthridine's reduction potential could not be determined due to solvent decomposition in the respective voltage range (see ESI†). However, it can be assumed to be more negative than the reduction potential of the Togni reagent, because no cathodic reduction was detected in a solution of phenanthridine 3a in acetonitrile (0.1 M Bu4NPF6) below −2 V vs. Fc/Fc+. Sustaining the radical chain is therefore feasible.
Initial reactions were performed under conditions similar to the established ones2 by reacting 2-isocyano-1,1′-biphenyl 1a with 1.5 equiv. of the Togni reagent 2 at 80 °C in dioxane. Tetrabutylammonium hexafluorophosphate was added to improve the conductivity (see ESI† for a detailed description of the experimental setup). The electrolysis was performed at a constant current of 0.12 mA until an overall electric charge of 0.075 faraday per mol was reached and the desired 6-(trifluoromethyl)phenanthridine 3a was obtained in 66% yield (Table 1, entry 2). In a control experiment without electrochemical initiation product 3a was formed in traces only (Table 1, entry 1). Hence, an injected electron induced more than 8 catalytic cycles before termination of the chain reaction occurred, clearly documenting its catalytic character. Herein, the exclusive cathodic reduction of the Togni reagent is assumed. Practically, a fraction of the current might also be consumed by other components in the reaction mixture implying an actually higher number of turnovers.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 2 (equiv.) | No. of faraday per mol | Solvent (mM) | Temp. (°C) | Yielda (%) |
| a Yield determined by 19F-NMR using trifluorotoluene as an internal standard (reactions run with 0.2 mmol of 1a). b Bu4NBF4 (0.1 M) as supporting electrolyte. c Constant current of 0.01 mA. d Reaction performed under air. e I2 (12 mol%) were added. | |||||
| 1 | 1.5 | 0.00 | Dioxane (80) | 80 | 3 |
| 2 | 1.5 | 0.075 | Dioxane (80) | 80 | 66 |
| 3 | 1.5 | 0.075 | Dioxane (40) | 80 | 17 |
| 4 | 1.5 | 0.075 | Dioxane (160) | 80 | 61 |
| 5 | 1.5 | 0.075 | Dioxane (80) | 80 | 65b |
| 6 | 1.5 | 0.007 | Dioxane (80) | 80 | 6c |
| 7 | 1.5 | 0.075 | Dioxane (80) | 80 | 56d |
| 8 | 1.5 | 0.11 | Dioxane (80) | 80 | 70 |
| 9 | 1.5 | 0.075 | Dioxane (80) | 70 | 71 |
| 10 | 1.5 | 0.075 | Dioxane (80) | 60 | 6 |
| 11 | 1.5 | 0.11 | Dioxane (80) | 70 | 69 |
| 12 | 2.0 | 0.075 | Dioxane (80) | 80 | 77 |
| 13 | 2.0 | 0.075 | Dioxane (80) | 70 | 53 |
| 14 | 2.0 | 0.0050 | Dioxane (80) | 80 | 5 |
| 15 | 2.0 | 0.0075 | Dioxane (80) | 80 | 23 |
| 16 | 2.0 | 0.010 | Dioxane (80) | 80 | 49 |
| 17 | 2.0 | 0.025 | Dioxane (80) | 80 | 64 |
| 18 | 2.0 | 0.0075 | Dioxane (80) | 80 | 58e |
| 19 | 2.0 | 0.010 | Dioxane (80) | 80 | 65e |
| 20 | 2.0 | 0.075 | Dioxane (80) | 80 | 66e |
Further reaction optimization was performed by variation of the concentration (Table 1, entries 3 and 4) and by using tetrabutylammonium tetrafluoroborate as supporting electrolyte (Table 1, entry 5), but the yield could not be improved. Decreasing the charge quantity to 0.007 faraday per mol did neither improve the yield nor the number of turnovers per electron (Table 1, entry 6). However, increasing the charge quantity to 0.11 faraday per mol afforded a slightly higher yield (70%, Table 1, entry 8). A similar effect on the yield was noted upon running the reaction at 70 °C (Table 1, entry 9). In this case, the increase of the charge quantity did not lead to an improved yield (Table 1, entry 11). At 60 °C, yield significantly decreased (Table 1, entry 10). The highest yield (77%), representing a turnover number of 10, could be obtained by increasing the equivalents of the Togni reagent to 2 at 80 °C (Table 1, entry 12). Decreasing the reaction temperature to 70 °C did not improve the yield (Table 1, entry 13).
Furthermore, we were interested in the number of turnovers at the onset of the reaction. Hence, we stopped the reaction after charge quantities of 0.0050, 0.0075, 0.010, and 0.025 faraday per mol were reached (Table 1, entries 14–17). Surprisingly, in the former case a low yield of 5% is observed. Increasing the overall electric charge to 0.0075 and 0.010 faraday per mol, however, leads to significantly increased yields of 23% and 49%, resembling high turnover numbers of about 30 and 49. The initial early termination of the chain reaction might be ascribed to the presence of inhibitors or the low concentration of formed ortho-iodobenzoate. Addition of iodine (12 mol%) as a redox mediator further improves the efficacy of the catalytic cycle at the early stage of the reaction and turnover numbers up to 77 are achieved (Table 1, entries 18 and 19). The yield could not be improved by this means (Table 1, entry 20).
With the optimized conditions in hand, we additionally explored the generality of the electrochemical initiation by varying the isonitrile component (Fig. 3). Trifluoromethylation of 4′-methyl and 4′-fluoro biarylisonitriles 1b and 1c yielded the corresponding phenanthridines 3b and 3c in slightly lower yields. 8-Chloro-6-(trifluoromethyl)phenanthridine 3d was obtained in 75% yield. Note that also 8-iodo-6-(trifluoromethyl)phenanthridine 3e could be prepared, despite intermediate formation of the corresponding radical anion that could also react via fragmentation of the C–I bond. Obviously, the intermolecular electron transfer to the Togni reagent is faster as compared to the iodide anion fragmentation. A high yield (79%) was also achieved for 2-fluoro-6-(trifluoromethyl)phenanthridine 3f. Various functional groups like cyanides (3g), ethers (3h), and esters (3i) are tolerated and the respective phenanthridines are obtained in satisfying yields of 72%, 69%, and 77%. A trimethylsilyl substituted phenanthridine (3j) could also be prepared in good yield (75%). The highest yield (80%) was observed for 2-methyl-6,8-bis(trifluoromethyl)phenanthridine (3k) revealing more than ten catalytic cycles per electron. 2-Methyl-6-(trifluoromethyl)phenanthridine 3l was obtained in 77% yield. Reaction of isonitrile 1m resulted in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of phenanthridines 3m and 3m′. Substituents in different positions of the isonitrile bearing arene moiety are also tolerated and 3- as well as 4-methyl-6-(trifluoromethyl)phenanthridine 3n and 3m were isolated in yields of 67% and 63%. The results suggest that various functional groups and substitution patterns are tolerated under the applied reaction conditions.
:1 mixture of phenanthridines 3m and 3m′. Substituents in different positions of the isonitrile bearing arene moiety are also tolerated and 3- as well as 4-methyl-6-(trifluoromethyl)phenanthridine 3n and 3m were isolated in yields of 67% and 63%. The results suggest that various functional groups and substitution patterns are tolerated under the applied reaction conditions.
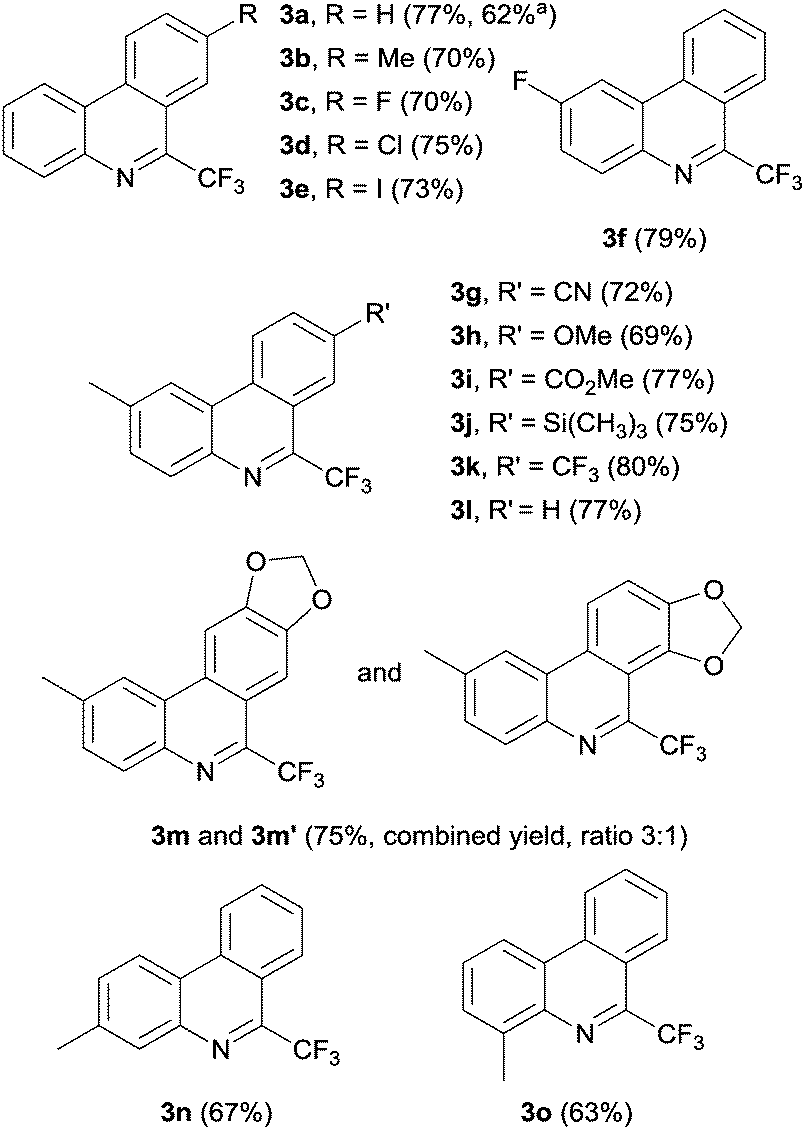 | ||
| Fig. 3 Various 6-trifluoromethylphenanthridines 3a–3f prepared under optimized conditions (isolated yield in bracket). a Reaction performed at 4 mmol scale (see ESI† for additional information). | ||
To examine the possibility of upscaling the cascade reaction, we performed the transformation at 4 mmol scale. Using a modified setup (non-optimized, see ESI† for additional information), phenanthridine 3a could be obtained in 62% yield by initiation with 0.035 faraday per mol of electrons.
In summary, 6-(trifluoromethyl)phenanthridines were prepared in good yields by electrochemically initiated trifluoromethylation of isonitriles. The total electric charge of 0.075 faraday per mol revealed the catalytic character of the electron, reaching turnover numbers up to 49. CV measurements support the suggested mechanism.
This work was supported by the Fonds der Chemischen Industrie (fellowship to M. L.) and the European Research Council (advanced grant agreement no. 692640).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS PubMed; (b) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed.
- (a) B. Zhang, C. Mück-Lichtenfeld, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2013, 52, 10792–10795 CrossRef CAS PubMed; (b) B. Zhang and A. Studer, Org. Lett., 2014, 16, 3990–3993 CrossRef CAS PubMed.
- (a) D. Leifert, C. G. Daniliuc and A. Studer, Org. Lett., 2013, 15, 6286–6289 CrossRef CAS PubMed; (b) B. Zhang and A. Studer, Org. Lett., 2014, 16, 1216–1219 CrossRef CAS PubMed; (c) B. Zhang and A. Studer, Org. Biomol. Chem., 2014, 12, 9895–9898 RSC; (d) D. Leifert, D. G. Artiukhin, J. Neugebauer, A. Galstyan, C. A. Strassert and A. Studer, Chem. Commun., 2016, 52, 5997–6000 RSC; (e) E. G. Mackay and A. Studer, Chem. – Eur. J., 2016, 22, 13455–13458 CrossRef CAS PubMed ; see also: ; (f) M. R. Torviso, D. Mansilla, S. Garcia, B. Lantaño, S. Barata-Vallejo and A. Postigo, J. Fluorine Chem., 2017, 197, 42–48 CrossRef CAS; (g) D. E. Yerien, R. Conde, S. Barata-Vallejo, B. Camps, B. Lantaño and A. Postigo, RSC Adv., 2016, 7, 266–274 RSC.
- (a) A. Dewanji, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2016, 55, 6749–6752 CrossRef CAS PubMed; (b) T. Hokamp, A. Dewanji, M. Lübbesmeyer, C. Mück-Lichtenfeld, E.-U. Würthwein and A. Studer, Angew. Chem., Int. Ed., 2017, 56, 13275–13278 CrossRef CAS PubMed.
- (a) S. Wertz, D. Leifert and A. Studer, Org. Lett., 2013, 15, 928–931 CrossRef CAS PubMed; (b) D. Leifert and A. Studer, Org. Lett., 2015, 17, 386–389 CrossRef CAS PubMed ; see also: ; (c) Z.-Q. Liu and Z. Li, Chem. Commun., 2016, 52, 14278–14281 RSC.
- (a) J. Xuan, C. G. Daniliuc and A. Studer, Org. Lett., 2016, 18, 6372–6375 CrossRef CAS PubMed; (b) J. Xuan, D. Gonzalez-Abradelo, C. A. Strassert, C. G. Daniliuc and A. Studer, Eur. J. Org. Chem., 2016, 4961–4964 CrossRef CAS; (c) X. Tang and A. Studer, Chem. Sci., 2017, 8, 6888–6892 RSC.
- Z.-Y. Gu, J.-J. Cao, S.-Y. Wang and S.-J. Ji, Chem. Sci., 2016, 7, 4067–4072 RSC.
- D. E. Yerien, S. Barata-Vallejo, B. Camps, A. E. Cristófalo, M. E. Cano, M. L. Uhrig and A. Postigo, Catal. Sci. Technol., 2017, 7, 2274–2282 CAS.
- C. Qu, Z. Wu, W. Li, H. Du and C. Zhu, Adv. Synth. Catal., 2017, 359, 1672–1677 CrossRef CAS.
- (a) J. Pinson and J. M. Saveant, J. Am. Chem. Soc., 1978, 100, 1506–1510 CrossRef CAS; (b) J. M. Saveant, Acc. Chem. Res., 1980, 13, 323–329 CrossRef CAS.
- (a) A. J. Bard, V. J. Puglisi, J. V. Kenkel and A. Lomax, Faraday Discuss. Chem. Soc., 1973, 56, 353–366 RSC; (b) G. Mabon and J. Simonet, Electrochim. Acta, 1992, 37, 2467–2468 CrossRef CAS; (c) A. Goulet-Hanssens, M. Utecht, D. Mutruc, E. Titov, J. Schwarz, L. Grubert, D. Bléger, P. Saalfrank and S. Hecht, J. Am. Chem. Soc., 2017, 139, 335–341 CrossRef CAS PubMed.
- C. Amatore, J. Badoz-Lambling, C. Bonnel-Huyghes, J. Pinson, J. M. Saveant and A. Thiebault, J. Am. Chem. Soc., 1982, 104, 1979–1986 CrossRef CAS.
- (a) J. Delaunay, G. Mabon, A. Orliac and J. Simonet, Tetrahedron Lett., 1990, 31, 667–668 CrossRef CAS; (b) Y. Roh, H.-Y. Jang, V. Lynch, N. L. Bauld and M. J. Krische, Org. Lett., 2002, 4, 611–613 CrossRef CAS PubMed.
- (a) S. D. Phillips and R. N. Castle, J. Heterocycl. Chem., 1981, 18, 223–232 CrossRef CAS; (b) T. Ishikawa, Med. Res. Rev., 2001, 21, 61–72 CrossRef CAS PubMed; (c) L.-M. Tumir, M. R. Stojković and I. Piantanida, Beilstein J. Org. Chem., 2014, 10, 2930–2954 CrossRef PubMed.
- (a) L. Sripada, J. A. Teske and A. Deiters, Org. Biomol. Chem., 2008, 6, 263–265 RSC; (b) Q. Wang, X. Dong, T. Xiao and L. Zhou, Org. Lett., 2013, 15, 4846–4849 CrossRef CAS PubMed; (c) Y. Cheng, H. Jiang, Y. Zhang and S. Yu, Org. Lett., 2013, 15, 5520–5523 CrossRef CAS PubMed; (d) W. Fu, M. Zhu, F. Xu, Y. Fu, C. Xu and D. Zou, RSC Adv., 2014, 4, 17226–17229 RSC; (e) M. Zhu, W. Fu, G. Zou, C. Xu and Z. Wang, J. Fluorine Chem., 2014, 163, 23–27 CrossRef CAS; (f) Y.-F. Wang, G. H. Lonca, M. Le Runigo and S. Chiba, Org. Lett., 2014, 16, 4272–4275 CrossRef CAS PubMed; (g) H. Jiang, X. An, K. Tong, T. Zheng, Y. Zhang and S. Yu, Angew. Chem., 2015, 127, 4127–4131 CrossRef; (h) X. Tang, S. Song, C. Liu, R. Zhu and B. Zhang, RSC Adv., 2015, 5, 76363–76367 RSC; (i) J. Rong, L. Deng, P. Tan, C. Ni, Y. Gu and J. Hu, Angew. Chem., Int. Ed., 2016, 55, 2743–2747 CrossRef CAS PubMed; (j) S. Feng, T. Li, C. Du, P. Chen, D. Song, J. Li, X. Xie and X. She, Chem. Commun., 2017, 53, 4585–4588 RSC; (k) Y.-R. Liu, H.-Y. Tu and X.-G. Zhang, Synthesis, 2015, 3460–3466 CAS; (l) R. Sakamoto, H. Kashiwagi, S. Selvakumar, S. A. Moteki and K. Maruoka, Org. Biomol. Chem., 2016, 14, 6417–6421 RSC; (m) M. Zhu, W. Fu, G. Zou, C. Xu and Z. Wang, J. Fluorine Chem., 2015, 180, 1–6 CrossRef CAS; (n) Z. Zhang, X. Tang and W. R. Dolbier, Org. Lett., 2015, 17, 4401–4403 CrossRef CAS PubMed; (o) S. Wang, W.-L. Jia, L. Wang and Q. Liu, Eur. J. Org. Chem., 2015, 6817–6821 CrossRef CAS; (p) J.-W. Gu and X. Zhang, Org. Lett., 2015, 17, 5384–5387 CrossRef CAS PubMed; (q) W. Wan, G. Ma, J. Li, Y. Chen, Q. Hu, M. Li, H. Jiang, H. Deng and J. Hao, Chem. Commun., 2016, 52, 1598–1601 RSC; (r) H. Zhao, Z. Liu, J. Song and H. Xu, Angew. Chem., 2017, 129, 12906 CrossRef ; for a review see: ; (s) A. V. Lygin and A. de Meijere, Angew. Chem., Int. Ed., 2010, 49, 9094–9124 CrossRef CAS PubMed; (t) B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505–3521 RSC.
- Y. Gorbanev, D. Leifert, A. Studer, D. O’Connell and V. Chechik, Chem. Commun., 2017, 53, 3685–3688 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data. See DOI: 10.1039/c7cc09302k |
| This journal is © The Royal Society of Chemistry 2018 |