
An enzymatic advance in nicotine cessation therapy†
Song
Xue,  ‡a
Marsida
Kallupi,
‡b
Lauren C.
Smith,
a
Pedro O.
Miranda,
a
Olivier
George
b
and
Kim D.
Janda
*ac
‡a
Marsida
Kallupi,
‡b
Lauren C.
Smith,
a
Pedro O.
Miranda,
a
Olivier
George
b
and
Kim D.
Janda
*ac
‡a
Marsida
Kallupi,
‡b
Lauren C.
Smith,
a
Pedro O.
Miranda,
a
Olivier
George
b
and
Kim D.
Janda
*ac
Abstract
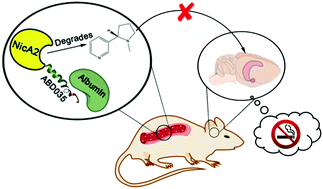
A nicotine-degrading enzyme termed NicA2 was altered (NicA2-J1) through fusion of an albumin binding domain to increase its half-life. Examination of NicA2-J1 in vivo demonstrated a complete blockade of brain nicotine access, which in turn blunted nicotine's psychoactive effects. These data further support development of pharmacokinetic nicotine cessation therapeutics.
 Please wait while we load your content...
Please wait while we load your content...

