
Barbiturate end-capped non-fullerene acceptors for organic solar cells: tuning acceptor energetics to suppress geminate recombination losses†
Ching-Hong
Tan,  a
Andrew
Wadsworth,
a
Sarah
Holliday,
*c
Samson A.
Jenekhe,
d
Derya
Baran,
e
Iain
McCulloch
ae
and
James R.
Durrant
*a
a
Andrew
Wadsworth,
a
Sarah
Holliday,
*c
Samson A.
Jenekhe,
d
Derya
Baran,
e
Iain
McCulloch
ae
and
James R.
Durrant
*a
a
Andrew
Wadsworth,
a
Sarah
Holliday,
*c
Samson A.
Jenekhe,
d
Derya
Baran,
e
Iain
McCulloch
ae
and
James R.
Durrant
*a
Abstract
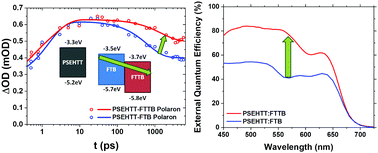
We report the synthesis of two barbiturate end-capped non-fullerene acceptors and demonstrate their efficient function in high voltage output organic solar cells. The acceptor with the lower LUMO level is shown to exhibit suppressed geminate recombination losses, resulting in enhanced photocurrent generation and higher overall device efficiency.
 Please wait while we load your content...
Please wait while we load your content...

