
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamic blue light-switchable protein patterns on giant unilamellar vesicles†
S. M.
Bartelt‡
a,
E.
Chervyachkova‡
a,
J.
Steinkühler
b,
J.
Ricken
a,
R.
Wieneke
 c,
R.
Tampé
c,
R.
Dimova
b and
S. V.
Wegner
*a
c,
R.
Tampé
c,
R.
Dimova
b and
S. V.
Wegner
*a
aMax Planck Institute for Polymer Research, 55128, Mainz, Germany. E-mail: wegners@mpip-mainz.mpg.de
bDepartment of Theory and Biosystems, Max Planck Institute of Colloids and Interfaces, 14476, Potsdam, Germany
cInstitut für Biochemie, Biozentrum, Cluster of Excellence Frankfurt, Goethe-Universität Frankfurt, 60438, Frankfurt, Germany
First published on 10th January 2018
Abstract
The blue light-dependent interaction between the proteins iLID and Nano allows recruiting and patterning proteins on GUV membranes, which thereby capture key features of patterns observed in nature. This photoswitchable protein interaction provides non-invasive, reversible and dynamic control over protein patterns of different sizes with high specificity and spatiotemporal resolution.
Protein patterns and local protein clustering are essential during many biological processes, such as cell signalling, division, and migration as well as during all stages of tissue formation and development.1,2 These patterns are highly dynamic and yet precisely regulated in space and time.3–5 It is of great interest to produce such dynamic protein patterns in vitro to understand and control the underlying processes, however our ability to do so is limited. Classical approaches to pattern molecules, such as lithography, micro-contact printing and chemical vapour deposition require multiple steps, harsh conditions (e.g. UV light, high temperature, non-physiological pH) and chemicals, which are not biocompatible.6–8
Light responsive approaches are particularly promising as visible light provides the desired high spatiotemporal control and is bioorthogonal. Visible and near infra-red light has been used to photopattern proteins by locally heating the thermoresponsive polymer poly(N-isopropylacrylamide)6 or decomposing ruthenium complexes with upconverting nanoparticles.9 Subsequently, the changes in surface chemistry allow proteins to unspecifically adsorb on the substrates following the photopattern. In these approaches all proteins in solution adsorb indiscriminately and the protein integrity is not assured. Photocleavable nitrobenzyl caging groups are useful to photopattern proteins through specific interactions. Nitrobenzyl groups have been used to control the interaction between Ni2+-NTA (N-nitrilotriacetic acid) groups and His-tagged proteins,10,11 biotin and streptavidin12 as well as glutathione and glutathione S-transferase.13 In particular, photocaged lipids allow recruiting proteins with high spatial and temporal control to phospholipid membranes, which are important cell models to study protein function.8,14 Yet, the decaging of nitrobenzyl groups is irreversible, which does not allow to alter protein patterns reversibly and requires cytotoxic UV light.15 Reversible protein photopatterns can only be produced with azobenzenes, which change surface properties when they undergo cis–trans isomerization.16,17 However, the unspecificity of the protein interaction, the requirement of UV light for the trans to cis isomerization, and in particular the photodynamic equilibrium limit this approach. Overall, there is still a need for a highly specific, biocompatible, and reversible way to pattern proteins with the desired spatiotemporal control that operates in the presence of other biomolecules under physiological conditions.
To achieve this goal, we suggest using photoswitchable protein heterodimers recently developed in the field of optogenetics. In particular, we utilized the proteins iLID (improved light-inducible dimer, based on the photoswitchable LOV2 domain from Avena sativa) and Nano (wild-type SspB), which specifically interact with each other under blue light (488 nm) and dissociate from each other in the dark. These proteins have been used to reversibly control various processes in live cells.18,19 In particular, iLID with a membrane anchoring tag has been expressed in cells to localize intracellular Nano fused proteins to the cell membrane under blue light. In this study, we anchor purified iLID to the outer membrane of giant GUVs (giant unilamellar vesicles) and recruit proteins fused to Nano to the outer GUV membrane in situ with blue light (Fig. 1a). Since the binding of Nano to iLID is reversible in the dark, we will be able to form reversible and dynamic protein patterns with precise control in space and time. The high specificity of the iLID/Nano interaction and the response to blue light will allow patterning a specific protein in the presence of other biomolecules including lipids and without damaging them.
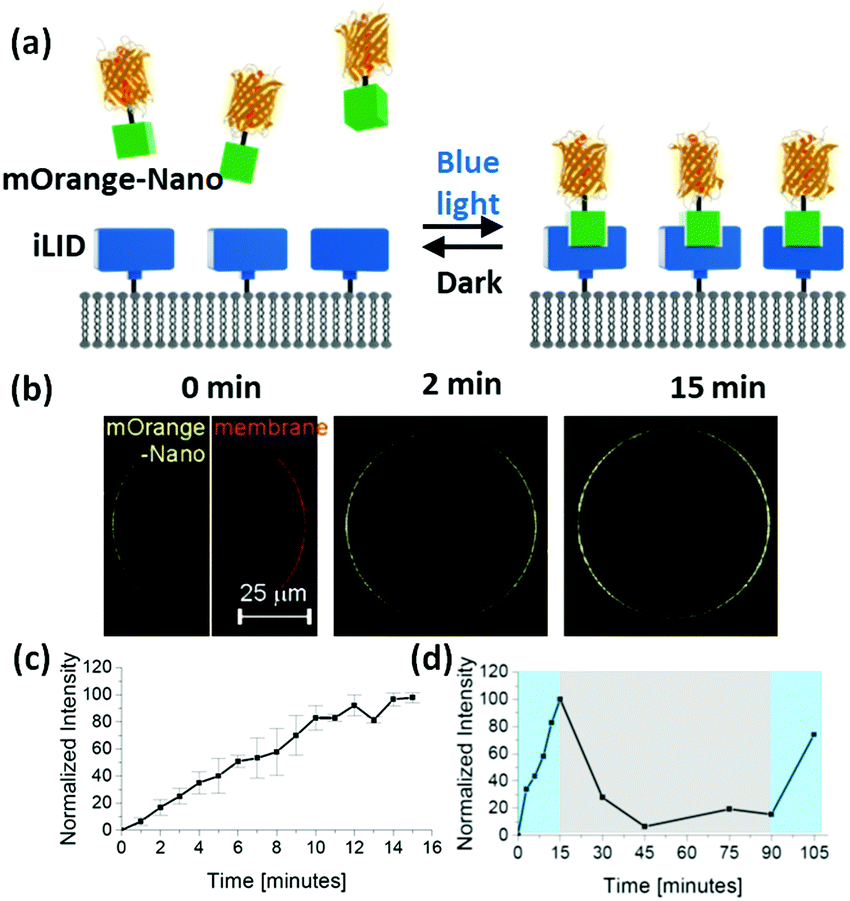 | ||
| Fig. 1 (a) Schematic representation of mOrange-Nano recruitment to iLID-functionalized lipid bilayer under blue light but not in the dark. (b) Fluorescence images of iLID-decorated GUVs (red) in the presence of mOrange-Nano (yellow) under blue light illumination (5% laser power: 575.5 ± 21 nW, 488 nm). (c) mOrange-Nano recruitment to GUV membrane under blue light. Error bars show the standard deviation from 3 independent experiments. (d) Change in fluorescence intensity under blue light (coloured in blue) and in the dark (coloured in grey). | ||
To demonstrate this concept, we immobilized His6-tagged iLID on GUVs with Ni2+-NTA groups on their surface (lipid composition: POPC with 10 mol% POPG + 0.1 mol% DGS-NTA-Ni2+ + 1 mol% DiD dye). When we transferred these GUVs into a solution of mOrange-Nano (orange fluorescent protein fused to Nano), upon blue light illumination over 15 min we could observe a gradual increase in mOrange fluorescence on the GUV membrane (Fig. 1b). We quantified this increase by comparing the fluorescence intensity on the membrane before and after illumination (Fig. 1c). Subsequently, when we placed the GUVs in the dark, the fluorescence intensity at the membrane decreased, proving that the recruitment of the mOrange-Nano is reversible. In the dark after 15 min approximately 70% and after 30 min approximately 95% of the recruited mOrange-Nano had dissociated from the iLID-decorated GUVs (Fig. 1d). Additionally, mOrange-Nano can be recruited to the GUVs multiple times. After renewed blue light illumination for 15 min, the fluorescence at the GUV membrane reached the same level as after the first blue light illumination. Hence, the photoswitchable interaction between iLID and Nano can be used to reversibly and repeatedly recruit proteins fused to Nano onto an iLID-decorated GUV. This interaction can in principle also be used to pattern on different types of surfaces. As an example, we have immobilized iLID on glass surfaces coated with Ni2+-NTA terminated PEG (polyethylene glycol) chains. Also in these samples we observed more mOrange-Nano recruitment to the surface under blue light illumination than in the dark (Fig. S1, ESI†).
The amount of the protein that recruits to a membrane is one of the key parameters to control protein activity. Using the iLID/Nano interaction, we can easily adjust how much protein recruits to the GUV through the intensity of the blue light (Argon laser, 488 nm). Already light powers as low as 70 nW through a 63× water objective, which corresponds to 1% of the laser power, are sufficient to partially activate iLID and recruit mOrange-Nano to the membrane (Fig. 2a). Higher light powers up to 6.3 μW (20% laser power) increased both the rate and the amount of protein recruitment. Further, intensity increase led to no additional recruitment. This can either be due to the full activation of iLID achieving the photostationary state or the bleaching of the fluorescent protein.
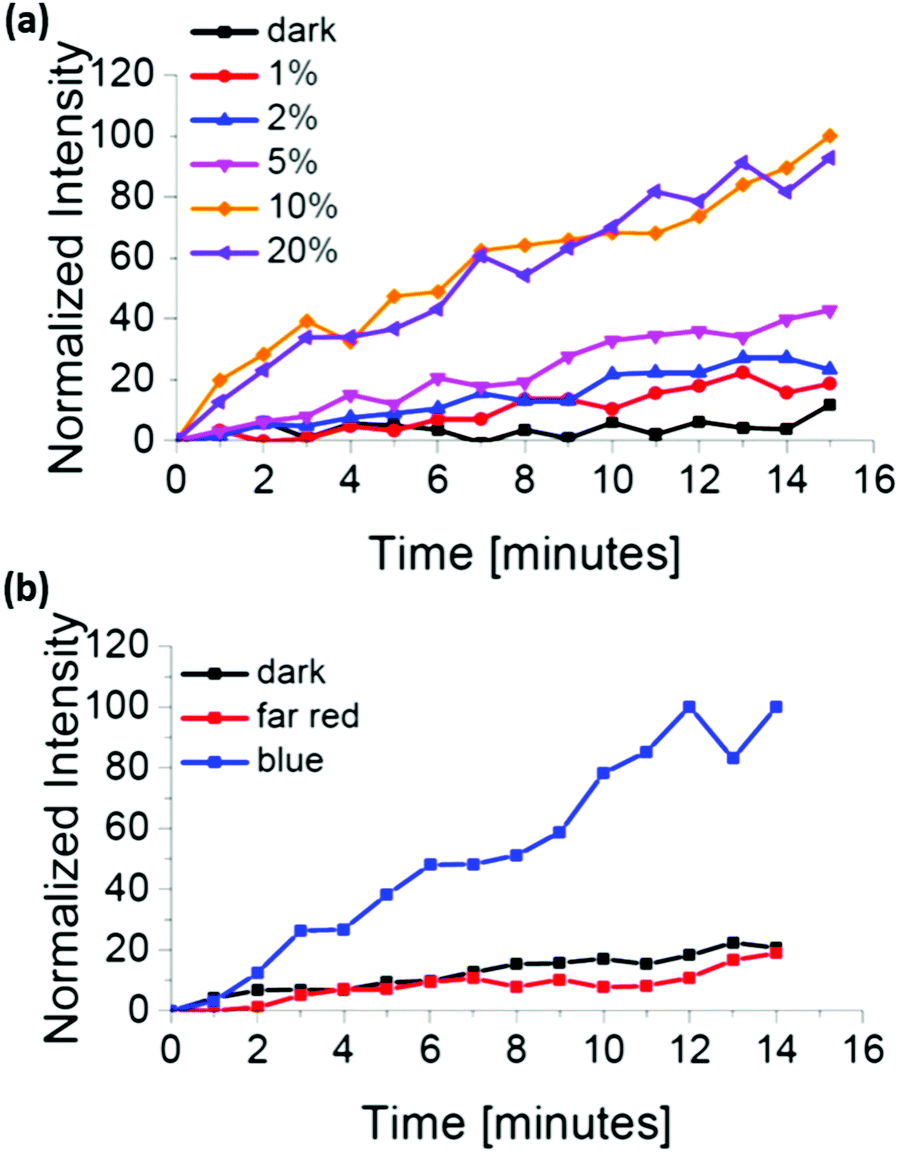 | ||
| Fig. 2 mOrange-Nano recruitment to iLID-functionalized GUVs depending on (a) the blue light intensity and (b) the illumination wavelengths. | ||
We also investigated if the iLID/Nano interaction is selectively induced exclusively under blue light and not with light of different wavelengths. In the absence of blue light illumination, we observed that mOrange-Nano is not recruited to the iLID-modified GUV membrane (Fig. 2b). During our experiments, we excited mOrange with green light (561 nm) for fluorescence imaging, and prepared the samples under red light (633 nm) (labelled as dark). Similarly, far-red (750 nm) light illumination fails to induce mOrange-Nano recruitment to the membrane. The lack of protein recruitment in the absence of blue light illumination, despite the use of green, red and far-red light, confirms the selectivity for blue light. The orthogonality to other wavelengths allows combining the iLID/Nano protein interactions with red light-dependent protein interactions to specifically pattern two different proteins, as it has been achieved in optogenetic studies.20,21
We further quantified the changes in kinetic and thermodynamic parameters for immobilized iLID and Nano under blue light and in the dark. The reasons to utilize the QCM-D (Quartz Crystal Microbalance with Dissipation Monitoring) for these measurements are twofold. First, QCM-D allows to measure protein surface interactions with high sensitivity in real time. Second, it is a non-spectral technique, so that the measuring process does not interfere with the light sensitive proteins and allows performing the measurements in either complete darkness or under blue light. For the measurements we immobilized His6-tagged iLID on supported lipid bilayers (lipid composition: DOPC + 5 mol% DGS-NTA) and subsequently titrated increasing concentrations of Nano on these surfaces under blue light or in the dark (Fig. 3 and Fig. S2, ESI†). From decrease in frequency, which is proportional to the amount of protein bound to the surface, it is evident that Nano binds to immobilized iLID better under blue light than in the dark. For example, already at 500 nM Nano efficiently binds to iLID under blue light but there is very little binding at the same concentration in the dark. Yet, it should be noted that the two proteins interact under both conditions. We modelled the QCM-D data to obtain the thickness of the adsorbed protein layer and to calculate Kd (dissociation constant) as well as kon and koff (association and dissociation rate constant, respectively) under blue light and in the dark (Table 1 and Fig. S3, ESI†). From the modelling we derived the Kd values for the interaction of Nano with immobilized iLID to be 280 nM under blue light and 1.2 μM in dark (Table 1). This equals more than 4-fold difference in binding affinity between the lit state and the dark. In the literature the affinity between the two proteins in solution changes from 132 nM under blue light to 4.7 μM in the dark, which is a 36-fold change.19 This shows, that the immobilization of iLID on a lipid membrane significantly influences the light dependent change in affinity to Nano. Investigating the recruitment of mOrange-Nano to GUVs with different lipid composition (DOPC vs. POPC) and fluidity (POPC + 20–40 mol% cholesterol), we observe that these parameters do not affect mOrange-Nano binding under blue light (Fig. S4, ESI†). Hence, we propose that the steric hindrance imposed by the immobilization of iLID results in a lower change in affinity from the dark to the lit state. As in many optogenetic studies one interaction partner is localized to a membrane this effect is of major importance.19,20
 | ||
| Fig. 3 Thermodynamic study of the iLID/Nano interaction (a) in the dark and (b) under the blue light using QCM-D. Increasing concentrations of Nano are binding to an iLID-functionalized supported lipid bilayer. | ||
| Constants | Blue light | Dark |
|---|---|---|
| K d [μM] | 0.28 ± 0.14 | 1.20 ± 0.40 |
| k off [min−1] | 5.4 × 10−2 ± 0.8 × 10−2 | 11.0 × 10−2 ± 5.0 × 10−2 |
| k on [μM−1 s−1] | 3.3 × 10−3 ± 0.8 × 10−3 | 1.6 × 10−3 ± 0.7 × 10−3 |
The koff can be computed from the washing off of Nano from the surface in the QCM-D measurements. The calculated koff values are 5.4 × 10−2 min−1 (half-life 12.8 min) and 11.0 × 10−2 min−1 (half-life 6.3 min) under blue light and in the dark, respectively (Table 1). The dynamic range of the protein unbinding is on the order of minutes to hours, which is in agreement with our findings of enrichment and depletion of proteins to the GUV membrane (Fig. 1c and d). Also in literature, the interaction between iLID and Nano is reported to reverse within minutes in optogenetic studies.19 The affinity range of nano- to micromolar and the time range of minutes, which the iLID/Nano interaction can cover to form dynamic protein patterns, are highly relevant to the affinity and time scale observed for protein patterns in nature.22,23
We sought to use the high spatiotemporal control that the iLID/Nano interaction provides to locally control protein enrichment on a GUV membrane. Towards this end, we illuminated a small region of interest (ROI) on one side of an iLID-decorated GUV, while continuously imaging to observe mOrange-Nano recruitment (Fig. 4a). Despite the high fluidity of the lipid membrane, the fluorescence intensity increases locally in the ROI (Fig. 4b). For a more quantitative analysis, we compared the local fluorescence increase in the ROI to the mirrored area on the other side of the GUV. The illuminated ROI showed consistently higher fluorescence intensity compared to the area on the opposite side of the GUV (Fig. S5, ESI†). The local illumination over time led to gradual increase in fluorescence over the whole GUV. The high lipid membrane fluidity leads to diffusion of the recruited protein from the ROI at one pole of the GUV and allows forming transient protein gradients on the GUV. Presumably, fixed patterns can be achieved on membranes in the gel phase where diffusion is hindered, but the biological relevance of gel-phase membranes is limited.
 | ||
| Fig. 4 Local recruitment of mOrange-Nano to a ROI on a single GUV. Fluorescence images of iLID-decorated GUVs in the presence of mOrange-Nano before (a) and after (b) local illumination with blue light (488 nm Argon Laser, 15 min) at the ROI (yellow rectangle). Scale bar is 15 μm. | ||
We also are able to spatially control the recruitment of proteins to one specific GUV in the presence of another. To achieve this, we have locally illuminated only one iLID-decorated GUV with blue light in the presence of another GUV (Fig. 5a and b). Indeed, we observe recruitment of mOrange-Nano just to the illuminated GUV but not the other. During tissue formation protein patterns emerge over many cells and we would like to mimic this by recruiting proteins at the scale of multiple GUVs.24 As a tissue-like substrate, we created a carpet of GUVs (lipid composition: POPC + 10 mol% POGP + 1 mol% trisNTA-Suc-DODA25) on a PVA (polyvinyl alcohol) substrate so that the GUVs remained supported by it and had limited mobility (Fig. 5c and d). As in previous results, we could recruit mOrange-Nano to a whole carpet of GUVs under blue light illumination within minutes (Fig. 5e) and reverse it (Fig. S6, ESI†). By only illuminating a ROI on the GUV carpet, we locally recruited mOrange-Nano onto multiple GUVs (Fig. 5f). Interestingly, the membrane fluidity also leads to protein recruitment to connected membranes of neighbouring GUVs. Additionally, we demonstrate that depending on the size of the ROI, the protein recruitment can range from a few to many GUVs (Fig. S7, ESI†). Overall, it is possible to sequentially pattern proteins and to dynamically reverse these patterns according to the needs with great flexibility in size and shape.
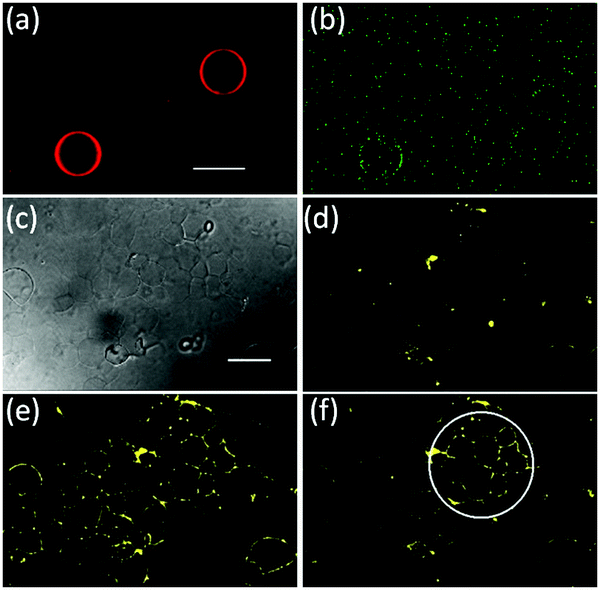 | ||
| Fig. 5 Selective GUV activation. (a) Fluorescent image of two GUVs. Scale bar 25 μm. (b) Local mOrange-Nano recruitment to a single GUV by local blue light illumination. (c–f) Patterning of mOrange-Nano on an iLID-functionalized GUV carpet. (c) DIC image of the immobilized GUVs. Fluorescence images of the GUV carpet (d) in the dark, (e) fully activated with blue light and (f) locally activated in a ROI with blue light (488 nm) for 1 min. The circle approximately indicates the irradiated region. Scale bar 25 μm. | ||
In summary, the photoswitchable interaction between iLID and Nano can be used to photopattern proteins with blue light. Unlike existing protein patterning methods, the iLID/Nano interaction is reversible and dynamic, non-invasive, operates under physiological conditions and is very specific, which allows using it in complex environments with multiple biomolecules. Most importantly, the protein patterns can be formed with high spatial and temporal resolution in affinity and time scales relevant to biology. The detailed characterization of thermodynamic and kinetic parameters for iLID/Nano provides insight into the protein interaction, reversion and pattern dynamics. In this study we broaden the application of iLID and Nano from optogenetics, where they are used to control cellular processes, to photopattern proteins on materials. Here, we have focused on protein recruitment onto lipid bilayers as these mimic the physiological surrounding proteins operate in. However, this is not limited to lipid interphases and extendable to any substrate functionalized with Ni2+-NTA to immobilize the His6-tagged iLID and to any protein of interest fused to Nano. This patterning approach is also scalable from the level of a single GUV to the level of a GUV carpet providing exceptional versatility. As the protein pair iLID/Nano only responds to blue light, it would be possible to combine it with other light responsive interactions to simultaneously pattern multiple proteins with high complexity and yet exquisite control.18–21
This work is part of the MaxSynBio consortium, which is jointly funded by the Federal Ministry of Education and Research (BMBF) (FKZ 031A359L) of Germany and the Max Planck Society (MPG). E.C. would like to thank the MPG for a doctoral fellowship. The pQE-80L iLID (C530M) and pQE-80L MBP-SspB Nano (Addgene plasmid # 60408 and 60409) were a gift from Brian Kuhlman. Our thanks go to Dr. Raimund Sauter for help with fitting the QCM-D data, Simge Yüz for her insightful comments, and to Stefan Schumacher for the illustrations. Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.References
- N. C. Hartman and J. T. Groves, Curr. Opin. Cell Biol., 2011, 23, 370 CrossRef CAS PubMed.
- J. Schweizer, M. Loose, M. Bonny, K. Kruse, I. Monch and P. Schwille, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15283 CrossRef CAS PubMed.
- S. Kretschmer and P. Schwille, Curr. Opin. Cell Biol., 2016, 38, 52 CrossRef CAS PubMed.
- C. A. DeForest and D. A. Tirrell, Nat. Mater., 2015, 14, 523 CrossRef CAS PubMed.
- P. Dillard, F. Pi, A. C. Lellouch, L. Limozin and K. Sengupta, Integr. Biol., 2016, 8, 287 RSC.
- C. Reuther, R. Tucker, L. Ionov and S. Diez, Nano Lett., 2014, 14, 4050 CrossRef CAS PubMed.
- L. Filipponi, P. Livingston, O. Kaspar, V. Tokarova and D. V. Nicolau, Biomed. Microdevices, 2016, 18, 1 CrossRef CAS PubMed.
- A. K. Rudd, J. M. Valls Cuevas and N. K. Devaraj, J. Am. Chem. Soc., 2015, 137, 4884 CrossRef CAS PubMed.
- Z. Chen, S. He, H. J. Butt and S. Wu, Adv. Mater., 2015, 27, 2203 CrossRef CAS PubMed.
- N. Labòria, R. Wieneke and R. Tampé, Angew. Chem., Int. Ed., 2013, 14(52), 848 CrossRef PubMed.
- S. V. Wegner, O. I. Sentürk and J. P. Spatz, Sci. Rep., 2015, 5, 18309 CrossRef CAS PubMed.
- T. Terai, E. Maki, S. Sugiyama, Y. Takahashi, H. Matsumura, Y. Mori and T. Nagano, Chem. Biol., 2011, 18, 1261 CrossRef CAS PubMed.
- V. Gatterdam, R. Ramadass, T. Stoess, M. A. Fichte, J. Wachtveitl, A. Heckel and R. Tampe, Angew. Chem., Int. Ed., 2014, 53, 5680 CrossRef CAS PubMed.
- X. Chen, M. Venkatachalapathy, D. Kamps, S. Weigel, R. Kumar, M. Orlich, R. Garrecht, M. Hirtz, C. M. Niemeyer, Y.-W. Wu and L. Dehmelt, Angew. Chem., Int. Ed., 2017, 56, 5916 CrossRef CAS PubMed.
- R. B. Setlow, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3363 CrossRef CAS.
- J. Zhang, T. Hu, Y. Liu, Y. Ma, J. Dong, L. Xu, Y. Zheng, H. Yang and G. Wang, ChemPhysChem, 2012, 13, 2671 CrossRef CAS PubMed.
- D. Pearson and A. D. Abell, Chem. – Eur. J., 2010, 16, 6983 CrossRef CAS PubMed.
- O. I. Lungu, R. A. Hallett, E. J. Choi, M. J. Aiken, K. M. Hahn and B. Kuhlman, Chem. Biol., 2012, 19, 507 CrossRef CAS PubMed.
- G. Guntas, R. A. Hallett, S. P. Zimmerman, T. Williams, H. Yumerefendi, J. E. Bear and B. Kuhlman, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 112 CrossRef CAS PubMed.
- M. J. Kennedy, R. M. Hughes, L. A. Peteya, J. W. Schwartz, M. D. Ehlers and C. L. Tucker, Nat. Methods, 2010, 7, 973 CrossRef CAS PubMed.
- K. Müller, R. Engesser, S. Metzger, S. Schulz, M. M. Kämpf, M. Busacker, T. Steinberg, P. Tomakid, M. Ehrbar, F. Nagy, J. Timmer, M. D. Zubriggen and W. Weber, Nucleic Acids Res., 2013, 41, 7 CrossRef PubMed.
- F. Wu, J. Halatek, M. Reiter, E. Kingma, E. Frey and C. Dekker, Mol. Syst. Biol., 2016, 12, 873 CrossRef PubMed.
- S. K. Saka, A. Honigmann, C. Eggeling, S. W. Hell, T. Lang and S. O. Rizzoli, Nat. Commun., 2014, 5, 4509 CAS.
- J. Hao, B. Zou, K. Narayanan and A. George, Bone, 2004, 34, 921 CrossRef CAS PubMed.
- S. Lata, A. Reichel, R. Brock, R. Tampé and J. Piehler, J. Am. Chem. Soc., 2005, 127, 10205 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc08758f |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2018 |