

Abstract
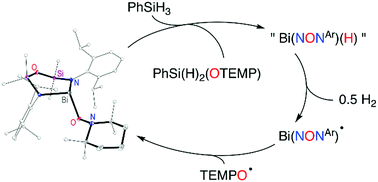
Bismuth(III) TEMPOxide compounds have been synthesized from the coupling of Bi(II) species with the TEMPO˙ radical. The steric profile of the supporting bis(amido)disiloxane ligand promotes different fluxional behaviour in solution, and DFT calculations suggest variation in the Bi–O bond character. These compounds are active catalysts for oxidative coupling of TEMPO and silane substrates, believed to proceed via metathesis of Bi–O and Si–H bonds followed by decomposition of bismuth-hydride intermediate species.
- This article is part of the themed collection: Philip Power at 65: an icon of organometallic chemistry
 Please wait while we load your content...
Please wait while we load your content...

