

Abstract
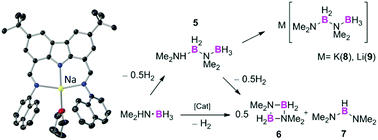
Group 1 salts containing carbazolido NNN pincer ligands are precatalysts for the dehydrogenation of Me2NH·BH3. NMR monitoring and DOSY studies show a heavy dependence on the metal and solvent, allowing in some cases selective formation of dehydrogenation products consistent with hydrogen liberation.
- This article is part of the themed collection: Philip Power at 65: an icon of organometallic chemistry
 Please wait while we load your content...
Please wait while we load your content...

