
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible modification of DNA by methyltransferase-catalyzed transfer and light-triggered removal of photo-caging groups†
Lea
Anhäuser
a,
Fabian
Muttach
a and
Andrea
Rentmeister
 *ab
*ab
aUniversity of Münster, Department of Chemistry, Institute of Biochemistry, Wilhelm-Klemm-Str. 2, 48149 Münster, Germany. E-mail: a.rentmeister@uni-muenster.de
bCells-in-Motion Cluster of Excellence (EXC1003-CiM), University of Münster, Germany
First published on 24th November 2017
Abstract
Methyltransferases are powerful tools for site-specific transfer of non-natural functional groups from synthetic analogs of their cosubstrate S-adenosyl-L-methionine (AdoMet). We present a new class of AdoMet analogs containing photo-caging (PC) groups in their side chain, enzymatic transfer of PC groups by a promiscuous DNA MTase as well as light-triggered removal from the target DNA. This strategy provides a new avenue to reversibly modulate the functionality of DNA at MTase target sites.
Methyltransferases (MTases) modify several classes of biomolecules post-synthetically and modulate their biological function. In many cases, methylation is dynamic and reversible, indicating a regulatory function, as well documented for histone methylation in epigenetics.1,2 For DNA, the importance of reversible methylation (m5C) for controlling transcriptional activity has recently emerged.3,4 In mRNA, the most abundant internal modification was found to be a reversible methylation (m6A) with functional implications in mRNA metabolism and translation.5
The cosubstrate S-adenosyl-L-methionine (AdoMet) serves as nature's methyl donor and synthetic analogs with extended allylic and propargylic groups have proven useful for transferring numerous functional side-chains to specific sites of MTase substrates. The powerful combination of promiscuous or engineered MTases and AdoMet analogs led to a new method for site-specific derivatization of DNA,6,7 RNA,8–13 proteins,7,14,15 and small molecules.16 Another class of AdoMet analogs, in which the amino acid side chain is replaced by an aziridine, enabled the transfer of the entire cosubstrate.17 By extending the adenine at the 8 position, transfer of biotin or fluorescent labels directly to DNA was achieved.17,18 This approach was applied to optically map individual, long DNA molecules and DNA-binding proteins18 as well as to identify bacteriophage strain types.19 In the field of RNA, AdoMet analogs were used for fluorescent labeling of tRNA and mRNA but also the transfer of photo-crosslinkers and affinity labels was reported.8,9,13,20–22
However, while methylation in nature is often a regulatory process that can be reverted, reversibility of the enzymatically introduced non-natural groups has not been reported, impeding our abilities to study the important dynamic aspects of enzymatic modifications and the downstream processes. On the other hand, light is orthogonal to most biological processes and can be used as trigger to remove photo-caging (PC) groups.23 Oligonucleotides with PC groups have opened the field of optochemical biology, providing a new level of spatio-temporal control in processes at all levels of gene expression, as exemplified by photo-caged siRNAs, plasmids, triplex forming oligonucleotides and aptamers.23,24 To date, PC groups are usually installed during chemical synthesis of oligonucleotides and require a combination with ligation strategies to access large nucleic acid molecules such as plasmids in order to bring them under control of light.25
Inspired by the enormous potential of optochemical biology, we thought that MTase-catalyzed introduction of PC groups might provide a straightforward route to modify biomacromolecules post-synthetically and reversibly. We therefore designed and synthesized AdoMet analogs for the enzymatic transfer of typical PC groups. Specifically, we appended the o-(ONB) and p-nitrobenzyl-(PNB), the 4-acetyl-2-nitrobenzyl-(ANB), and the 2-anthraquinonyl-residues (ANT) to S-adenosyl-L-homocysteine, yielding AdoMet analogs 1a–d, termed AdoONB, AdoPNB, AdoANB and AdoANT, respectively (Fig. 1A). For synthesis of the four AdoPCs, S-adenosyl-L-homocysteine was reacted with the corresponding bromides, which were synthesized as described previously (Scheme S1, Fig. S1 and S2, ESI†).26,27 All of those AdoMet analogs contain aromatic ring systems as side-chains, which are bulky and might be incompatible with several MTases. However, our recent work on RNA- and DNA-MTases demonstrated that (i) many highly promiscuous MTases are suitable for this purpose and (ii) the benzyl group promotes enzymatic transfer by promiscuous MTases.28 In fact, the PNB group was more efficiently transferred from AdoPNB than the methyl group from AdoMet by the guanine N7 methyltransferase from the microsporidian parasite Encephalitozoon cuniculi (relative specificity of >300%).28
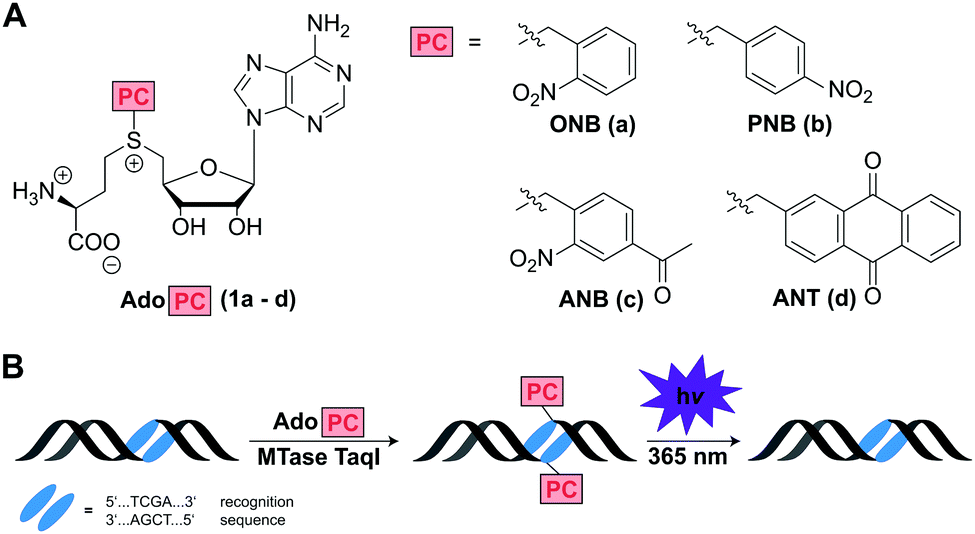 | ||
| Fig. 1 (A) AdoMet analogs designed for enzymatic transfer and light-triggered removal of functional groups to MTase targets. PC: photo-caging group. (B) Concept of MTase-catalyzed transfer of a photo-cleavable moiety from AdoPC to dsDNA and subsequent photo-cleavage. 2′-Deoxyadenosine of the indicated recognition sequence is modified at the N6 position by M. TaqI. | ||
Based on this work and literature on DNA MTases6,7,18,29 we anticipated that 1a–d would be efficiently converted by the highly promiscuous DNA MTase TaqI, thus protecting the recognition site from enzymatic cleavage by the corresponding restriction enzyme R. TaqI. Irradiation by light (365 nm) should remove the ONB-,30,31 ANB-,27 and ANT-group32,33 resulting in the first reversible, non-natural modification of DNA that can be enzymatically installed (Fig. 1B).
To test whether 1a–d are accepted by promiscuous MTases, we used a short dsDNA substrate, M. TaqI and 1a–d, followed by enzymatic degradation to single nucleosides and LC–MS-based analysis. In all cases, the N6-modified 2′-deoxyadenosine (2a–d) was observed, indicating successful enzymatic installation of the respective PC-group (Fig. 2B and Fig. S7–S9, ESI†).
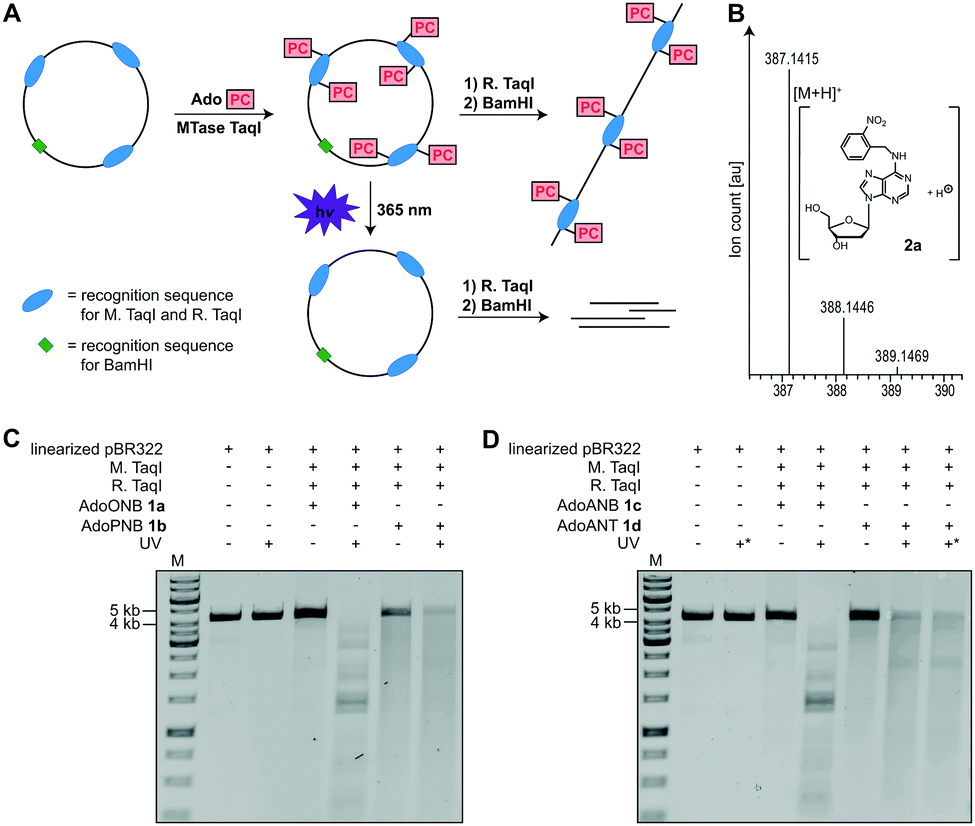 | ||
| Fig. 2 Enzymatic modification of DNA plasmid pBR322 using MTase TaqI and subsequent photo-cleavage. (A) Concept for the transfer of AdoPC to the N6 position of 2′-deoxyadenosine using MTase TaqI followed by irradiation with UV light (λmax = 365 nm). For clarity, only three of the seven M. TaqI recognition sites in pBR322 are shown. (B) Mass spectrometric analysis of N6-ONB modified 2′-deoxyadenosine 2a. A DNA oligonucleotide was subjected to enzymatic PC-modification, followed by digestion using nuclease P1 and dephosphorylation using FastAP. Calculated mass of [C17H19N6O5]+ = 387.1411 [M + H]+, found 387.1415. (C and D) Analysis of enzymatic modification and photo-cleavage of plasmid DNA according to A. Conditions: irradiation at 365 nm for 10 min or 30 min (marked with *), 1% agarose gel (100 V, 1 h), staining with ethidium bromide. M: GeneRuler™ 1 kb DNA ladder (Thermo Fisher Scientific). | ||
Next, we used plasmid DNA (pBR322) containing seven M. TaqI recognition sites and carried out enzymatic modification with 1a–d, followed by enzymatic digest using R. TaqI and plasmid linearization (Fig. 2A). Similar to modification with natural AdoMet, modification using 1a–d protected pBR322 from degradation by R. TaqI, indicating that all seven recognition sites are efficiently modified, with relative efficiencies AdoMet ≫ AdoANT > AdoPNB > AdoONB > AdoANB (Fig. 2C, D and Fig. S5, S6, ESI†).
To test whether the PC-groups can be removed by light, the enzymatically modified plasmids were irradiated at 365 nm using an LED followed by R. TaqI digestion. We observed that 1a-treated samples containing the ONB-group at the N6 of 2′-deoxyadenosine were cleaved by R. TaqI after 10 minutes of UV irradiation (Fig. 2C), suggesting that the ONB-group was removed. A control showing unmodified pBR322 plasmid after UV irradiation demonstrated that degradation is not the result of the UV irradiation itself. The PNB-group however was not photo-cleavable, as 1b-treated plasmid did not become susceptible to R. TaqI degradation after UV-irradiation (Fig. 2C). The ANB-group could also be removed by light, as plasmid treated with 1c became susceptible to R. TaqI-based restriction after irradiation at 365 nm for 10 min (Fig. 2D). Results obtained with the ANT-group were ambiguous: irradiation of plasmid treated with 1d decreased the amount of full-length DNA. However, the restriction products were longer than expected for complete cleavage – even when the irradiation time was extended to 30 min – suggesting that not all of the ANT-groups were removed.
These data indicate that the ONB-group and the ANB-group can be efficiently removed from the N6 position of 2′-deoxyadenosine by irradiation at 365 nm, in line with previous reports where the 1-(o-nitrophenyl)-ethyl (NPE) group was synthetically installed at the N6 position of 2′-deoxyadenosine.34,35 The PNB-group appears to be unsuitable as photo-caging group, as irradiation only decreased the amount of full-length DNA, but without leading to defined shorter bands in line with previous reports.36 The ANT-group has been reported as PC-group for alcohols,32 aldehydes,2,37 ketones,2 carboxylic acids33 and cAMP.38 When appended to the N6 of 2′-deoxyadenosine we observed only partial removal. Nevertheless, the ANT-group might become also useful for this purpose if substitutions facilitating cleavage are found.
In summary, we present the first enzymatic strategy for reversible post-synthetic modification of DNA. Based on our previous observation that the benzyl group is a privileged moiety for MTase-catalyzed transfer, we developed a new class of AdoMet analogs containing PC-groups in their side-chain and demonstrated transfer to MTase-target sites of DNA. As expected, PC groups block enzymatic cleavage of plasmid DNA by restriction enzymes. Upon irradiation with light, two of these groups – namely the o-nitrobenzyl- and the 4-acetyl-2-nitrobenzyl groups – are efficiently removed and the DNA is again accessible for restriction enzymes. By combining enzymatic transfer, synthetic cosubstrates and photo-caging groups, our strategy presents a new avenue for the reversible modulation of biological functions of DNA that can be readily extended to other bio-macromolecules such as RNA and proteins.
We gratefully acknowledge funding by the DFG (RE2796/6-1, EXC 1003 Cells in Motion – Cluster of Excellence) and the Fonds der Chemischen Industrie (doctoral fellowship to L. A. and Dozentenpreis to A. R.). We thank Dr W. Dörner, A.-M. Lawrence-Dörner and S. Wulff for experimental assistance.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. J. Klose and Y. Zhang, Nat. Rev. Mol. Cell Biol., 2007, 8, 307 CrossRef CAS PubMed.
- J.-Y. Yu, W.-J. Tang, H.-B. Wang and Q.-H. Song, J. Photochem. Photobiol., A, 2007, 185, 101 CrossRef CAS.
- S. Schiesser, B. Hackner, T. Pfaffeneder, M. Müller, C. Hagemeier, M. Truss and T. Carell, Angew. Chem., Int. Ed., 2012, 51, 6516 CrossRef CAS PubMed.
- M. Münzel, D. Globisch and T. Carell, Angew. Chem., Int. Ed., 2011, 50, 6460 CrossRef PubMed.
- B. X. S. Zhao, I. A. Roundtree and C. He, Nat. Rev. Mol. Cell Biol., 2017, 18, 31 CrossRef CAS PubMed.
- C. Dalhoff, G. Lukinavicius, S. Klimasauskas and E. Weinhold, Nat. Chem. Biol., 2006, 2, 31 CrossRef CAS PubMed.
- G. Lukinavičius, V. Lapienė, Z. Staševskij, C. Dalhoff, E. Weinhold and S. Klimašauskas, J. Am. Chem. Soc., 2007, 129, 2758 CrossRef PubMed.
- Y. Motorin, J. Burhenne, R. Teimer, K. Koynov, S. Willnow, E. Weinhold and M. Helm, Nucleic Acids Res., 2011, 39, 1943 CrossRef CAS PubMed.
- M. Tomkuviene, B. Clouet-d'Orval, I. Cerniauskas, E. Weinhold and S. Klimasauskas, Nucleic Acids Res., 2012, 40, 6765 CrossRef CAS PubMed.
- J. M. Holstein, D. Stummer and A. Rentmeister, Protein Eng., Des. Sel., 2015, 28, 179 CrossRef CAS PubMed.
- J. M. Holstein, D. Stummer and A. Rentmeister, Chem. Sci., 2015, 6, 1362 RSC.
- J. M. Holstein, D. Schulz and A. Rentmeister, Chem. Commun., 2014, 50, 4478 RSC.
- D. Schulz, J. M. Holstein and A. Rentmeister, Angew. Chem., Int. Ed., 2013, 52, 7874 CrossRef CAS PubMed.
- K. Islam, W. Zheng, H. Yu, H. Deng and M. Luo, ACS Chem. Biol., 2011, 6, 679 CrossRef CAS PubMed.
- K. Islam, I. Bothwell, Y. Chen, C. Sengelaub, R. Wang, H. Deng and M. Luo, J. Am. Chem. Soc., 2012, 134, 5909 CrossRef CAS PubMed.
- S. Singh, J. J. Zhang, T. D. Huber, M. Sunkara, K. Hurley, R. D. Goff, G. J. Wang, W. Zhang, C. M. Liu, J. Rohr, S. G. Van Lanen, A. J. Morris and J. S. Thorson, Angew. Chem., Int. Ed., 2014, 53, 3965 CrossRef CAS PubMed.
- G. Pljevaljčić, F. Schmidt and E. Weinhold, ChemBioChem, 2004, 5, 265 CrossRef PubMed.
- S. Kim, A. Gottfried, R. R. Lin, T. Dertinger, A. S. Kim, S. Chung, R. A. Colyer, E. Weinhold, S. Weiss and Y. Ebenstein, Angew. Chem., Int. Ed., 2012, 51, 3578 CrossRef CAS PubMed.
- A. Grunwald, M. Dahan, A. Giesbertz, A. Nilsson, L. K. Nyberg, E. Weinhold, T. Ambjornsson, F. Westerlund and Y. Ebenstein, Nucleic Acids Res., 2015, 43, e117 CrossRef PubMed.
- F. Muttach, F. Mäsing, A. Studer and A. Rentmeister, Chem. – Eur. J., 2017, 23, 5988 CrossRef CAS PubMed.
- J. M. Holstein, F. Muttach, S. H. H. Schiefelbein and A. Rentmeister, Chem. – Eur. J., 2017, 23, 6165 CrossRef CAS PubMed.
- J. M. Holstein, L. Anhäuser and A. Rentmeister, Angew. Chem., Int. Ed., 2016, 55, 10899 CrossRef CAS PubMed.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446 CrossRef CAS PubMed.
- Q. Y. Liu and A. Deiters, Acc. Chem. Res., 2014, 47, 45 CrossRef CAS PubMed.
- J. Hemphill, J. Govan, R. Uprety, M. Tsang and A. Deiters, J. Am. Chem. Soc., 2014, 136, 7152 CrossRef CAS PubMed.
- M. Su, J. Wang and X. Tang, Chem. – Eur. J., 2012, 18, 9628 CrossRef CAS PubMed.
- L. Kammari, T. Solomek, B. P. Ngoy, D. Heger and P. Klan, J. Am. Chem. Soc., 2010, 132, 11431 CrossRef CAS PubMed.
- F. Muttach, N. Muthmann, D. Reichert, L. Anhäuser and A. Rentmeister, Chem. Sci., 2017, 8, 7947 RSC.
- C. Dalhoff, G. Lukinavicius, S. Klimasauskas and E. Weinhold, Nat. Protoc., 2006, 1, 1879 CrossRef CAS PubMed.
- J. A. Barltrop, P. J. Plant and P. Schofield, Chem. Commun., 1966, 822 RSC.
- P. Klan, T. Solomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119 CrossRef CAS PubMed.
- T. Furuta, Y. Hirayama and M. Iwamura, Org. Lett., 2001, 3, 1809 CrossRef CAS PubMed.
- M.-G. Ren, N.-M. Bi, M. Mao and Q.-H. Song, J. Photochem. Photobiol., A, 2009, 204, 13 CrossRef CAS.
- M. C. Buff, F. Schäfer, B. Wulffen, J. Müller, B. Pötzsch, A. Heckel and G. Mayer, Nucleic Acids Res., 2010, 38, 2111 CrossRef CAS PubMed.
- F. Schäfer, K. B. Joshi, M. A. Fichte, T. Mack, J. Wachtveitl and A. Heckel, Org. Lett., 2011, 13, 1450 CrossRef PubMed.
- P. Wan and K. Yates, Can. J. Chem., 1986, 64, 2076 CrossRef CAS.
- R. G. Brinson and P. B. Jones, Org. Lett., 2004, 6, 3767 CrossRef CAS PubMed.
- T. Furuta, H. Torigai, M. Sugimoto and M. Iwamura, J. Org. Chem., 1995, 60, 3953 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Biochemical and synthetic procedures, HPLC and mass spectrometric data. See DOI: 10.1039/c7cc08300a |
| This journal is © The Royal Society of Chemistry 2018 |