
A general and facile strategy for precisely controlling the crystal size of monodispersed metal–organic frameworks via separating the nucleation and growth†
Tiefeng
Wang  *a
*a
*a
Abstract
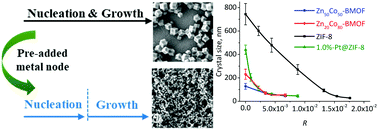
A novel and facile strategy was proposed for precisely controlling the crystal size of ZIF-8, ZnCo bimetallic ZIFs and also NPs@ZIF-8 by separating the nucleation and growth. The fundamentals of crystallization of this strategy were elaborately investigated, and the size effect on the internal diffusion was explored for a series of Pt@ZIF-8 catalysts.
 Please wait while we load your content...
Please wait while we load your content...

