
Near infrared imaging of Mer tyrosine kinase (MERTK) using MERi-SiR reveals tumor associated macrophage uptake in metastatic disease†
Miles A.
Miller
 *a,
Eunha
Kim
b,
Michael F.
Cuccarese
a,
Alec L.
Plotkin
a,
Mark
Prytyskach
a,
Rainer H.
Kohler
a,
Mikael J.
Pittet
a and
Ralph
Weissleder
*a
*a,
Eunha
Kim
b,
Michael F.
Cuccarese
a,
Alec L.
Plotkin
a,
Mark
Prytyskach
a,
Rainer H.
Kohler
a,
Mikael J.
Pittet
a and
Ralph
Weissleder
*a
aCenter for Systems Biology, Massachusetts General Hospital, 185 Cambridge Street, Boston, MA 02114, USA. E-mail: miles.miller@mgh.harvard.edu; rweissleder@mgh.harvard.edu
bDepartment of Molecular Science and Technology, Ajou University, Suwon 16499, Korea
First published on 23rd November 2017
Abstract
The receptor tyrosine kinase Mer (MERTK) is a promising drug target in cancer, where it can influence the metastasis-promoting signaling of both tumor cells and immune cells alike; however, no small molecule probes currently exist to selectively image Mer. In this work, we design and synthesize a selective near-infrared fluorescent molecular probe of Mer (MERi-SiR). Confocal microscopy of metastases in mice reveals predominant probe accumulation in Mer-expressing tumor-associated macrophages.
MERTK is a member of the TAM (TYRO3, AXL, MERTK) family of receptor tyrosine kinases, which are single-pass transmembrane proteins that transduce extracellular signals to intracellular phospho-signaling pathways influencing cell survival, migration, differentiation, and phagocytic activity. Mer can drive pro-metastasis signaling in tumor cells and is over-expressed in a variety of cancer-types, including melanoma and colorectal cancer.1 Mer signaling can also influence tumor-associated leukocytes, including macrophages, to downregulate their anti-tumor inflammatory responses and to enhance tumor-promoting wound-healing responses. Thus, inhibition or genetic silencing of Mer in both cancer cells and leukocytes has been shown to effectively limit tumor growth and metastasis. Mer plays a physiological role in efferocytosis, which is the clearing of dying cell debris by phagocytic immune cells. MERTK genetic mutations are associated with the accumulation of such debris, especially in the retina, thus contributing to the degenerative eye disease retinitis pigmentosa. Mer activity in phagocytes mediates processes in infection and sterile inflammation, for instance during atherosclerosis.2
Several Mer-targeting drugs have reached clinical trials. Gene therapies aim to express functional Mer in patients with retinitis pigmentosa (NCT01482195). In cancer, orally available Axl/Mer kinase inhibitors are tested in patients with leukemias (NCT03176277; ONO-7475) and solid tumors (NCT02729298; TP-0903).3 Furthermore, other clinically tested or even FDA-approved kinase inhibitors have significant cross-reactivity towards Mer, including foretinib (reached Ph-II trials; Mer Kd = 0.3 nM), sunitinib (sutent®, Mer Kd = 26 nM), and crizotinib (xalkori®).
Heterogeneity of Mer expression across patients, its role in metastasis throughout the body, its presence across a variety of cell types including both tumor cells and immune cells, and its therapeutic transgenic expression in ophthalmic disease provide motivation for developing methods to non-invasively image it at cellular or subcellular resolution. To accomplish this, we developed an imaging probe based on UNC2025, a first-in-class small molecule kinase inhibitor designed to specifically inhibit Mer with translationally suitable pharmacokinetic properties (Fig. 1A).4 UNC2025 binds Mer with an IC50 of 0.74 nM, with roughly 20-fold specificity over the other TAM receptors, Axl (IC50 14 nM) and Tyro3 (IC50 = 17 nM), although cross-reactivity with Flt3 was noted (IC50 = 0.8 nM).4 This compound is well tolerated in pre-clinical models and exhibits good circulating half-life, oral availability, kinetic solubility, and low clearance.4 Furthermore, the compound is effective in xenograft models of leukemia5 and solid cancers.6
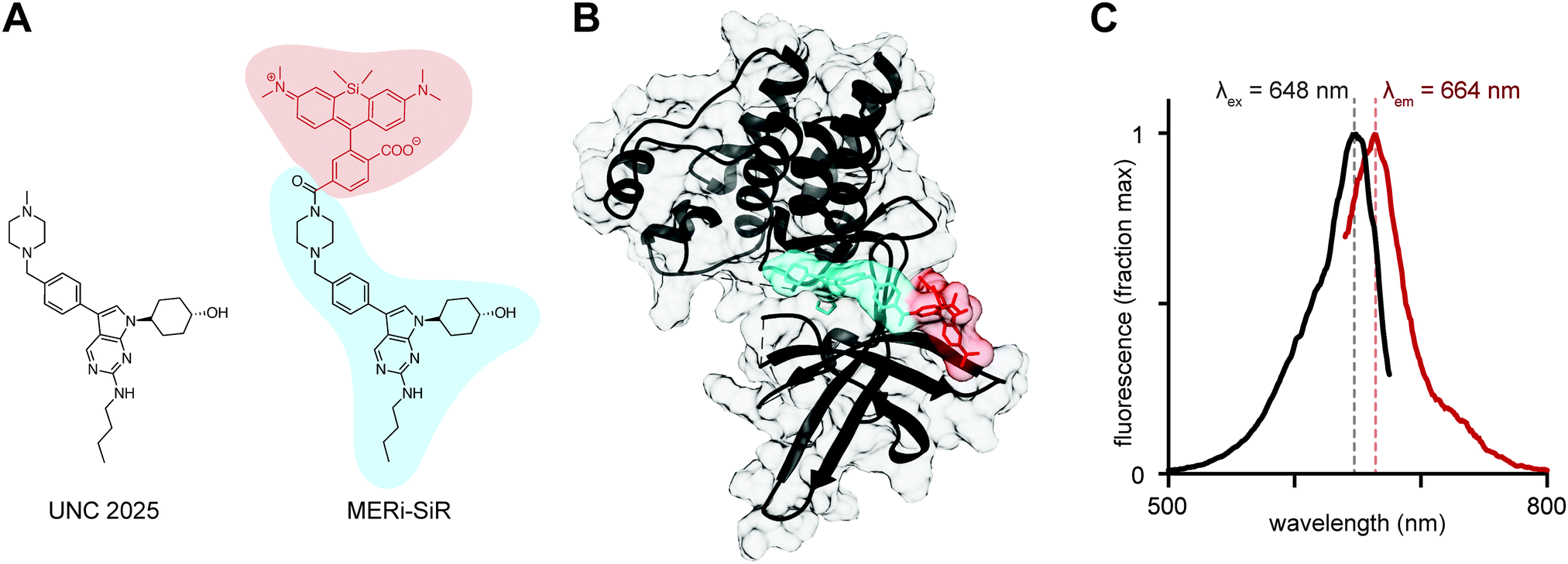 | ||
| Fig. 1 MERi-SiR structure and fluorescence spectra. (A) SiR-COOH (red) was conjugated to the Mer inhibitor UNC2025 (blue) to produce MERi-SiR. (B) Docking study of MERi-SiR with Mer, based on co-crystal structure of related compound UNC569; colors correspond to A. (C) Fluorescence excitation (black) and emission (red) spectra of MERi-SiR. | ||
We thus used UNC2025 as a scaffold to build a Mer-specific imaging probe, with silicon rhodamine carboxylate (SiR-COOH) as the fluorochrome.7 SiR-COOH is especially useful for small-molecule probes: it exhibits bright photostable fluorescence at near-infrared wavelengths, which are ideal for imaging deep through tissue; fluorescence is robust across physiological environments with little toxicity; it is amenable to super-resolution microscopy; and it exhibits an optimal degree of hydrophilicity for live-cell imaging.8
Known structure–activity relationships (SAR) of the pyrrolo[2,3-d]pyrimidine scaffold offered guidance on SiR-COOH conjugation to UNC2025. The trans-4-hydroxycyclohexyl group at the N1 position is important for Mer binding, with alternatives leading to attenuated potency or undesired hERG activity.9,10 The C6 position of the butylamine can be modified slightly, for example through substitution to cyclopropyl ethyl, but even this small change increases the IC50 of Mer binding by 1–2 fold.4 X-ray co-crystal structures of Mer with a closely related inhibitor (UNC569; PDB ID: 3TCP)9 indicate the C3 position is located away from the interior of Mer, towards the solvent, thus making this position the most promising candidate for fluorophore conjugation. The presence of a sulfonamide to bridge the N-methyl piperazine and the benzyl ring, employed in earlier analogues, as well as fluorination of the C3 position phenyl ring, had little effect on Mer inhibition.4
Based on SAR analysis, we designed a strategy to conjugate SiR-COOH to the C3 position N-methyl piperazine (Fig. 1A). MERi-SiR docking studies suggested the fluorochrome extends towards the solvent, thus preserving the compound's ability to access the binding pocket of the Mer active site (Fig. 1B). Based on this study, we first synthesized the N-tert-butyloxycarbonyl (N-Boc) protected-amine version of UNC2025, using slightly modified reported synthetic procedures.4 Amine deprotection and coupling of this compound to SiR-COOH yielded the Mer-inhibitor SiR-COOH conjugate (MERi-SiR) with an overall yield of 9.7% in 9 steps (Scheme S1, ESI†). Conjugation did not substantially affect the fluorescence spectra of SiR-COOH (Fig. 1C).
We tested the ability of MERi-SiR to visualize Mer in tissue culture, using SK-MEL-3 human melanoma cells, which overexpress Mer. Fluorescence imaging showed cytoplasmic accumulation of the probe (cytoplasmic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nuclear ratio of 2.0 ± 0.2:1), especially near the nucleus (Fig. 2A), which is consistent with the distribution of anti-Mer antibody staining in fixed and permeabilized cells (Fig. S1, ESI†), and with previously observed perinuclear and cytoplasmic Mer antibody-staining in permeabilized cancer cells that somewhat co-localized with the endoplasmic reticulum (http://www.proteinatlas.org). In contrast, perinuclear and cytoplasmic anti-Mer antibody accumulation was not observed in live-cell immunostaining conditions (Fig. S1, ESI†), suggesting that MERi-SiR substantially accumulates in intracellular pools of Mer that are inaccessible to the membrane-impermeant anti-Mer antibody. Co-incubation of MERi-SiR with excess UNC2025 decreases cellular fluorescence by 72% ± 9%, indicating the majority of accumulation is due to binding that can be competed off by the Mer-specific parent compound (Fig. 2B). We next used siRNA to reduce cellular expression of Mer and to test its impact on MERi-SiR fluorescence. SK-MEL-3 cells were again used, along with the murine macrophage cell line RAW264.7 as a model of Mer-expressing immune cells. Mer protein expression was quantified from cell lysate following treatment with Mer-targeting or control siRNA (Fig. 2C). Genetic knockdown reduced Mer by 45% and 55% in the melanoma and macrophage cultures, respectively (Fig. 2C). Encouragingly, MERi-SiR fluorescence also reduced by 41% and 53% in the melanoma and macrophage cultures, respectively, thus indicating correlation between protein content and MERi-SiR cellular retention.
:nuclear ratio of 2.0 ± 0.2:1), especially near the nucleus (Fig. 2A), which is consistent with the distribution of anti-Mer antibody staining in fixed and permeabilized cells (Fig. S1, ESI†), and with previously observed perinuclear and cytoplasmic Mer antibody-staining in permeabilized cancer cells that somewhat co-localized with the endoplasmic reticulum (http://www.proteinatlas.org). In contrast, perinuclear and cytoplasmic anti-Mer antibody accumulation was not observed in live-cell immunostaining conditions (Fig. S1, ESI†), suggesting that MERi-SiR substantially accumulates in intracellular pools of Mer that are inaccessible to the membrane-impermeant anti-Mer antibody. Co-incubation of MERi-SiR with excess UNC2025 decreases cellular fluorescence by 72% ± 9%, indicating the majority of accumulation is due to binding that can be competed off by the Mer-specific parent compound (Fig. 2B). We next used siRNA to reduce cellular expression of Mer and to test its impact on MERi-SiR fluorescence. SK-MEL-3 cells were again used, along with the murine macrophage cell line RAW264.7 as a model of Mer-expressing immune cells. Mer protein expression was quantified from cell lysate following treatment with Mer-targeting or control siRNA (Fig. 2C). Genetic knockdown reduced Mer by 45% and 55% in the melanoma and macrophage cultures, respectively (Fig. 2C). Encouragingly, MERi-SiR fluorescence also reduced by 41% and 53% in the melanoma and macrophage cultures, respectively, thus indicating correlation between protein content and MERi-SiR cellular retention.
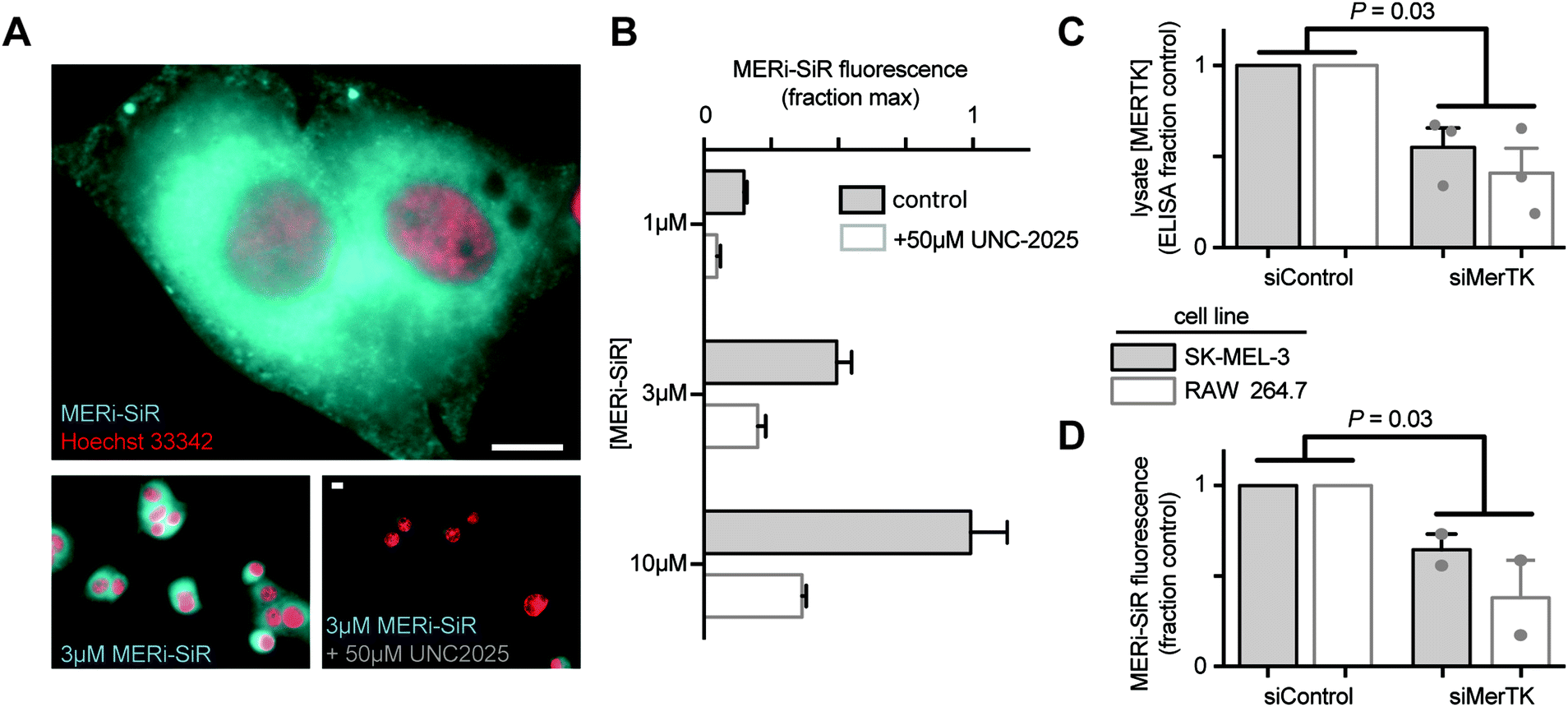 | ||
| Fig. 2 In vitro imaging analysis of MERi-SiR. (A and B) MERi-SiR distribution in SK-MEL-3 melanoma cells (A; scale bar 10 μm) and fluorescence intensity quantification (B; n > 50 cells) after 24 h incubation and subsequent washing, in the presence or absence of competing parent compound UNC2025. λex/λem for Hoechst and MERi-SiR were 360 nm/455 nm and 645 nm/705 nm, respectively, and values were normalized to the highest average cellular fluorescence observed across all conditions. (C and D) Quantification of reduced Mer expression following siRNA genetic knockdown (C) and corresponding reduction in MERi-SiR fluorescence intensity in the same cells (D). Data are means ± S.E.M., normalized to the average values observed in siControl conditions; p-values use two-tailed t-tests. | ||
To further test the activity of MERi-SiR towards Mer, we examined the effect of the probe on proliferation and phospho-signaling. As typical for small-molecule drug conjugates, MERi-SiR exhibited reduced cytotoxicity compared to the parent drug (Fig. 3A). This feature is actually beneficial from an imaging perspective, as it allows micromolar concentrations appropriate for fluorescence imaging to be used without causing widespread cell killing. In the same melanoma model, phosphorylation of Mer and the downstream kinase Erk1/2 (p44/42 MAPK) decreased by roughly 50% following MERi-SiR treatment at its IC50 concentration. Consistent with the cytotoxicity data, the parent compound exhibited more potent signaling inhibition (Fig. 3B and C). We examined whether exogenous addition of the Mer activating ligand, growth arrest specific 6 (Gas6), influenced the activity of MERi-SiR. Gas6 binds to the extracellular domain of Mer with nanomolar affinity and has the potential to stimulate conformational and phosphorylation changes of the receptor's cytoplasmic domain. However, in our experimental model, treatment with recombinant Gas6 had no discernible impact on constitutive Mer phospho-signaling or its inhibition by MERi-SiR, suggesting that exogenous Mer ligand does not prohibit MERi-SiR from interacting with its target (Fig. 3B and C). Taken together, these results suggest that MERi-SiR inhibits Mer signaling and cell growth, but less potently compared to unconjugated UNC2025.
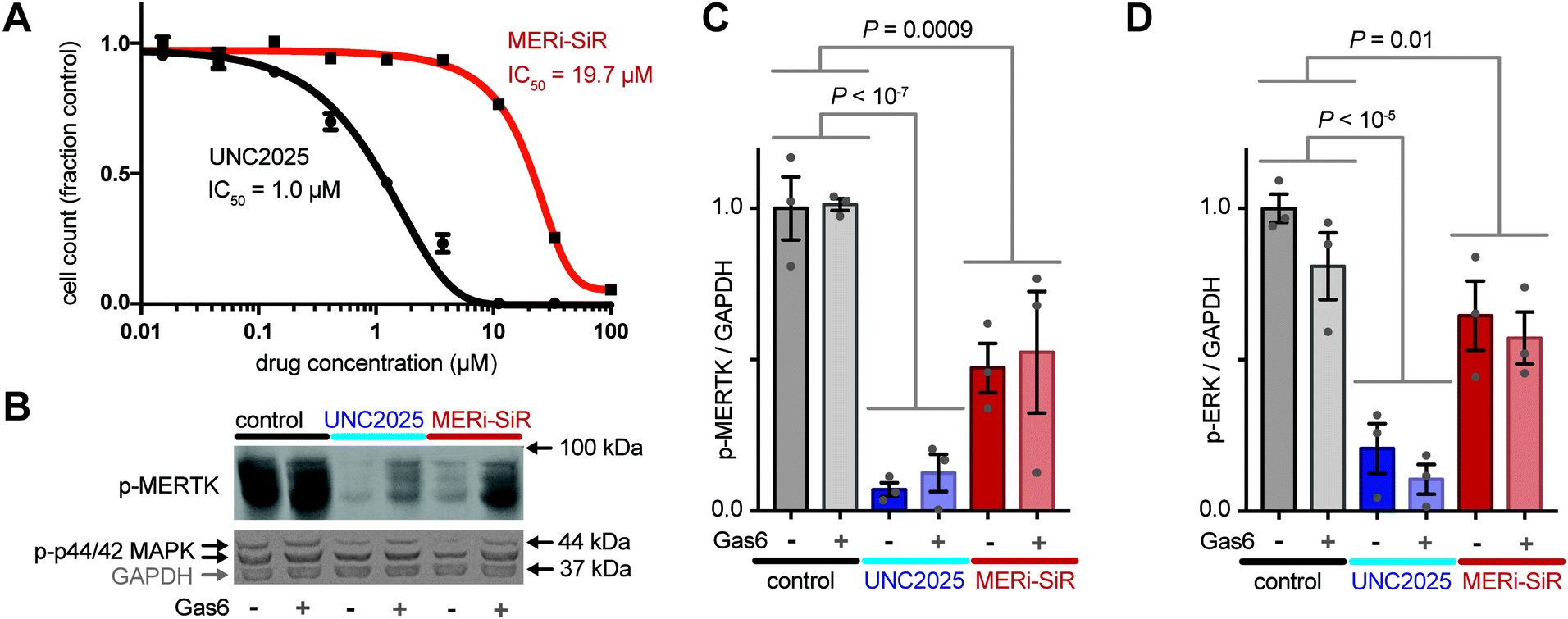 | ||
| Fig. 3 MERi-SiR inhibits Mer signaling, but with reduced potency. (A) Cell proliferation was measured after 72 h treatment. (B–D) Western blot (B) and quantification (C and D; normalized as fraction control) show decreased phosphorylation of Mer and downstream Erk1/2 (p44/42 MAPK) following treatment with UNC2025 or MERi-SiR. (A–D) Data are means ± S.E.M. using SK-MEL-3; p-values use two-tailed t-tests. | ||
We next tested whether MERi-SiR could selectively label Mer-expressing cells in vivo. We used a model of peritoneal carcinomatosis based on the implantation and subsequent metastatic spread of the murine colorectal cancer cell line CT26 to organs of the abdomen. To quantify specificity of MERi-SiR uptake in Mer-expressing cells, we used a genetically engineered CRISPR knock-in MERTKGFP/+ mouse model containing GFP+, Mer+ tumor associated macrophages.11 The genetically engineered mouse model does not use a Mer–GFP fusion protein; instead, GFP is expressed in the subset of host cells that endogenously express Mer (including tumor associated macrophages).11 Imaging revealed selective uptake of MERi-SiR in GFP+ cells across a variety of tissues, especially the skin and the spleen, with an average uptake in GFP+ cells roughly 4-fold that of GFP− cells (Fig. S2, ESI†). Similar patterns of selective uptake were also observed in the tumor stroma: MERi-SiR uptake was on average 4-fold higher in GFP+ cells than GFP− cells. Furthermore, cells containing more than 15-fold enhanced uptake of MERi-SiR were >95% GFP+, indicating specificity of the probe for Mer-expression at this high threshold level. Interestingly, although tumor cells exhibited a 40% average increase in MERi-SiR uptake compared to GFP− stroma (CT26 colorectal cancer cells express Mer12), this paled in comparison to the 400% increased MERi-SiR uptake in the GFP+ tumor-associated macrophages (Fig. 4). Across all organs and metastases (including peritoneal and splenic nodules), the highest MERi-SiR uptake was observed in metastatic nodules of the liver, which is the most common site of metastasis in colorectal cancer (Fig. S3, ESI†). Thus, these results suggest the importance of Mer and tumor associated macrophages in metastatic colorectal cancer, especially at sites in the liver.
 | ||
| Fig. 4 MERi-SiR accumulates in Mer+ tumor-associated-macrophages in a model of metastatic cancer. (A and B) Representative live-tissue confocal microscopy (A) of a metastasis (top) and corresponding zoomed-in details (bottom) show greatest MERi-SiR accumulation in Mer+ macrophages; corresponding quantification (B) of MERi-SiR uptake, with single-cell measurements shown as individual data points and their median ± 95% C.I. (n = 6 tumors). Scale bar 50 μm. λex/λem for GFP, tdTomato, and MERi-SiR were 473 nm/515 ± 25 nm, 559 nm/600 ± 22.5 nm, and 635 nm/705 ± 50 nm, respectively (± bandwidths). | ||
MERi-SiR provides a tool for selectively imaging Mer expression both in vitro and in vivo at subcellular resolution. The near-infrared SiR-COOH fluorochrome enables imaging deep through tissue, co-registration with commonly-used GFP and RFP reporters, and employs a fluorochrome compatible with super-resolution microscopy. Mer expression across multiple clinically-relevant tissues and cell-types have made fluorescent-protein based Mer reporters cumbersome in pre-clinical models; in contrast, MERi-SiR may have the potential in future work to be used across a spectrum of immunocompetent and patient-derived systems. In the clinic, MERi-SiR may be useful for optically monitoring Mer-based ophthalmic therapies, or for assessing Mer expression in viable tumor biopsies or patient-derived xenografts.
Full methodology details are described in the ESI.† All animal research was performed in accordance with the guidelines from the Institutional Subcommittee on Research Animal Care. Part of this work was supported by NIH/NCI grants K99CA207744, R01CA206890, and U01CA206997.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. T. Cummings, D. Deryckere, H. S. Earp and D. K. Graham, Clin. Cancer Res., 2013, 19, 5275 CrossRef CAS PubMed.
- B. Cai, E. B. Thorp, A. C. Doran, B. E. Sansbury, M. J. Daemen, B. Dorweiler, M. Spite, G. Fredman and I. Tabas, J. Clin. Invest., 2017, 127, 564 Search PubMed.
- A. Mollard, S. L. Warner, L. T. Call, M. L. Wade, J. J. Bearss, A. Verma, S. Sharma, H. Vankayalapati and D. J. Bearss, ACS Med. Chem. Lett., 2011, 2, 907 CrossRef CAS PubMed.
- W. Zhang, D. DeRyckere, D. Hunter, J. Liu, M. A. Stashko, K. A. Minson, C. T. Cummings, M. Lee, T. G. Glaros, D. L. Newton, S. Sather, D. Zhang, D. Kireev, W. P. Janzen, H. S. Earp, D. K. Graham, S. V. Frye and X. Wang, J. Med. Chem., 2014, 57, 7031 CrossRef CAS PubMed.
- D. DeRyckere, A. B. Lee-Sherick, M. G. Huey, A. A. Hill, J. W. Tyner, K. M. Jacobsen, L. S. Page, G. G. Kirkpatrick, F. Eryildiz, S. A. Montgomery, W. Zhang, X. Wang, S. V. Frye, H. S. Earp and D. K. Graham, Clin. Cancer Res., 2017, 23, 1481 CrossRef CAS PubMed.
- C. T. Cummings, W. Zhang, K. D. Davies, G. D. Kirkpatrick, D. Zhang, D. DeRyckere, X. Wang, S. V. Frye, H. S. Earp and D. K. Graham, Mol. Cancer Ther., 2015, 14, 2014 CrossRef CAS PubMed.
- G. Lukinavičius, K. Umezawa, N. Olivier, A. Honigmann, G. Yang, T. Plass, V. Mueller, L. Reymond, I. R. Corrêa, Z. G. Luo, C. Schultz, E. A. Lemke, P. Heppenstall, C. Eggeling, S. Manley and K. Johnsson, Nat. Chem., 2013, 5, 132 CrossRef PubMed.
- T. Ikeno, T. Nagano and K. Hanaoka, Chem. – Asian J., 2017, 12, 1435 CrossRef CAS PubMed.
- J. Liu, C. Yang, C. Simpson, D. Deryckere, A. Van Deusen, M. J. Miley, D. Kireev, J. Norris-Drouin, S. Sather, D. Hunter, V. K. Korboukh, H. S. Patel, W. P. Janzen, M. Machius, G. L. Johnson, H. S. Earp, D. K. Graham, S. V. Frye and X. Wang, ACS Med. Chem. Lett., 2012, 3, 129 CrossRef CAS PubMed.
- J. Liu, W. Zhang, M. A. Stashko, D. Deryckere, C. T. Cummings, D. Hunter, C. Yang, C. N. Jayakody, N. Cheng, C. Simpson, J. Norris-Drouin, S. Sather, D. Kireev, W. P. Janzen, H. S. Earp, D. K. Graham, S. V. Frye and X. Wang, Eur. J. Med. Chem., 2013, 65, 83 CrossRef CAS PubMed.
- M. A. Miller, R. Chandra, M. F. Cuccarese, C. Pfirschke, C. Engblom, S. Stapleton, U. Adhikary, R. H. Kohler, J. F. Mohan, M. J. Pittet and R. Weissleder, Sci. Transl. Med., 2017, 9, eaal0225 Search PubMed; J. F. Mohan, R. H. Kohler, J. A. Hill, R. Weissleder, D. Mathis and C. Benoist, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E7776 CrossRef CAS PubMed.
- R. Schmitz, A. F. Valls, R. Yerbes, S. von Richter, C. Kahlert, S. Loges, J. Weitz, M. Schneider, C. R. de Almodovar, A. Ulrich and T. Schmidt, Oncotarget, 2016, 7, 56355 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc07581b |
| This journal is © The Royal Society of Chemistry 2018 |