
Radical halogenation-mediated latent–active glycosylations of allyl glycosides†

Abstract
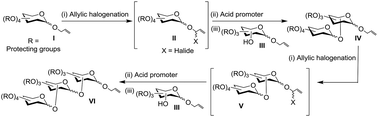
Radical halogenation-mediated glycosylation using allyl glycosides as donors and as acceptors emerges to be an efficient and hither-to unknown glycosylation method, adhering to the concept of the latent–active methodology. Several di- and trisaccharides that possess the allyl moiety at their reducing end are prepared through this new glycosylation methodology.
 Please wait while we load your content...
Please wait while we load your content...

