
Controlling the lectin recognition of glycopolymers via distance arrangement of sugar blocks†
K.
Jono
,
M.
Nagao
,
T.
Oh
,
S.
Sonoda
,
Y.
Hoshino
 and
Y.
Miura
*
and
Y.
Miura
*
Department of Chemical Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: miuray@chem-eng.kyushu-u.ac.jp
First published on 27th November 2017
Abstract
The arrangement of sugars in glycopolymers contributes to their recognition. The molecular recognition of proteins was controlled by the living radical polymerization of glycopolymers. The glycopolymers were prepared by the copolymerization of propargyl methacrylate (Pr-MA) and triethyleneglycol methacrylate (TEG-MA) via living radical polymerization with a reversible addition–fragmentation glycopolymer chain transfer (RAFT) reagent and by subsequent sugar conjugation by click chemistry. The block copolymers were prepared by the polymerization of Pr-MA and TEG-MA. The molecular recognition of glycopolymers was analyzed using the fluorescence quenching of lectin and found to be dependent on the glycopolymer structures. Two-site binding of glycopolymers to concanavalin A (ConA) was attained by both the glycopolymer with a 105-mer and the tri-block glycopolymer with a 103-mer. Glycopolymers with either a 27- or 54-mer showed much weaker interaction because of one-site binding. The molecular recognition of the glycopolymer was controlled by the arrangement and size of the sugar cluster and not by the sugar density.
The functions of organic molecules are determined by the functional groups and their stereo structures. A precise 3D structure is indispensable for functional organic materials, especially biopolymers. For example, the design of de novo peptides allows to obtain a particular arrangement of functional groups and a precise 3D structure;1 however, it is difficult to attain the precise stereo structure with synthetic polymers. Recently, synthetic polymers with complicated sequences have been reported using living radical polymerization.2,3 Multiblock polymers prepared via atomic transfer radical polymerization (ATRP),4,5 reversible addition fragmentation chain transfer (RAFT)6–9 and emulsion RAFT10 have been reported. The multiblock polymers can be prepared over 20 steps, as achieved with sequential synthesis of polypeptides.9,10 The major advance of sequential polymerization motivated us to fabricate precisely controlled functional materials such as de novo designed peptides.
On the other hand, one of the bio-functional synthetic polymers is a glycopolymer.11,12 Glycopolymers are polymers with pendant saccharides and exhibit strong molecular recognition to sugar recognition proteins. In this investigation, we controlled glycopolymer structures by controlling the polymerization of a RAFT reagent. In sugar–protein recognition, living radical polymerization facilitates the production of polymers with a complicated sequence. The preparation of multi-sequence polymers via living radical polymerization allows the precise arrangement of functional groups. Currently, glycopolymers are being studied on materials that exert multivalent effects.13 However, no research has been conducted to design and control molecular recognition by regulating the spatial arrangement of the chain glycopolymer, although trials were reported for biopolymers of polypeptides and DNA based on the precise 3D structure.14–16 Lectins have multiple ligand-binding sites with symmetric precise structures.17,18 For this reason, the spatial arrangement of the sugar is important in ensuring multivalent interactions with proteins. In this research, we designed the molecular recognition of glycopolymers via spatial arrangement of a sugar with block copolymers. The block polymer was prepared using RAFT living radical polymerization and the sugar–lectin interaction was analyzed via a fluorescence quenching measurement.19–21 Glycopolymers were polymerized with propargyl methacrylate (Pr-MA) and triethylene glycol monoethyl ether methacrylate (TEG-MA) as monomers, and mannose was conjugated via click chemistry.22 Three kinds of glycopolymers were synthesized: glycopolymers with a mannose side chain (H-series) based on the poly(Pr-MA) backbone (B-series), a di-block copolymer with poly(TEG-MA) (D) and a tri-block polymer composed of glycopolymer-b-poly(TEG-MA)-b-glycopolymer (T) (Fig. 1). The localization of the sugar–cluster was attained using the tri-block polymer and the molecular recognition was compared with that of the H polymers.
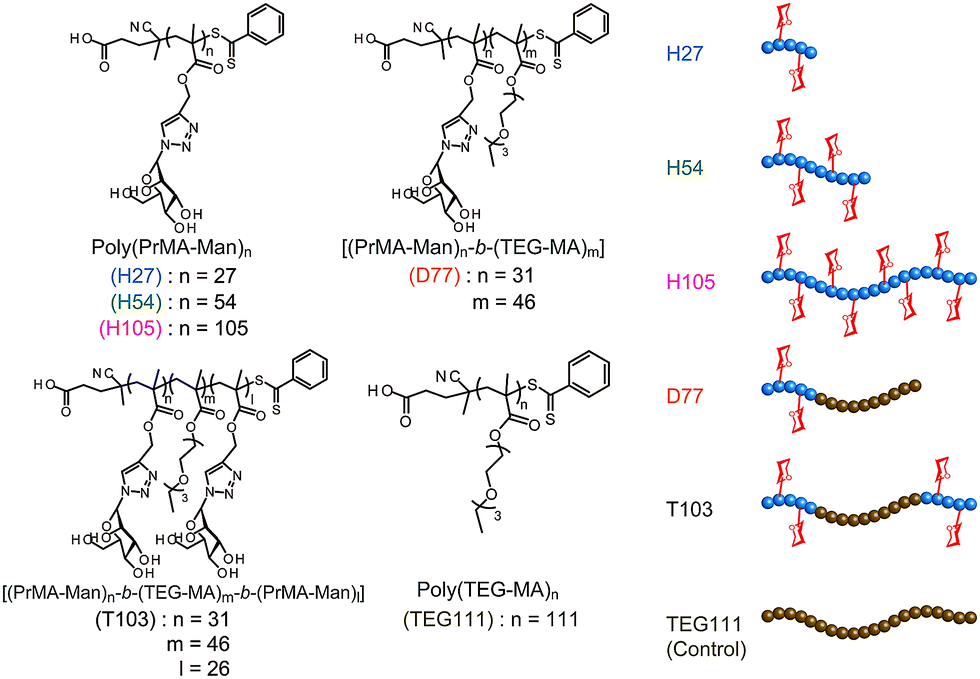 | ||
| Fig. 1 Chemical structures and schematic images of glycopolymers. | ||
A glycopolymer with different polymer chain lengths was prepared via living radical polymerization of poly(TMS-PrMA) (B-H27, B-H54 and B-H105, ESI†). Similar backbones of the di-block polymer (B-D77) and tri-block polymer (B-T103) were also prepared. Polymerization was performed in toluene at 60 °C using 4-cyano-4-(phenylcarbonothioylthio) pentanoic acid (CPADB) as a RAFT agent and azobisisobutyronitrile (AIBN) as an initiator. The degree of polymerization (DP) and molecular weights were confirmed by 1H-NMR and the Mw/Mn value by gel permeation chromatography (GPC) (Table S1, ESI†). The DP values determined by 1H-NMR were 27-, 54- and 105-mer, respectively, and the conversion of poly(Pr-MA) was over 90%. The Mw/Mn values of the polymers were low, suggesting the successful living radical polymerization, though the Mw/Mn value of B-T103 was relatively large (Fig. 2 and Table S1, ESI†). We also prepared the reference polymer of poly(TEG-MA) (TEG111) without the sugar unit as a control (Table 1).
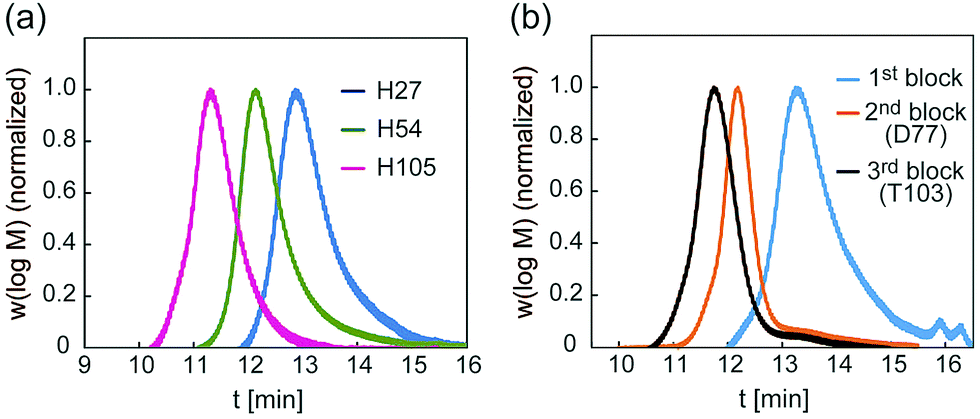 | ||
| Fig. 2 (a) GPC traces of backbones of H27, H54 and H105, and (b) GPC traces of backbones of D77 and T103. | ||
| 1st blocka (Pr-MA) (mer) | 2nd blocka (TEG-MA) (mer) | 3rd blocka (Pr-MA) (mer) | Sugar introductiona (%) | D.P. of sugar unita (mer) | M n,NMR (g mol−1) | M n,GPC (g mol−1) | M w/Mn | |
|---|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR. b Calculated by Pullulan standard. | ||||||||
| H27 | 27 | — | — | 100 | 27 | 9000 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 200 |
1.43 |
| H54 | 54 | — | — | 97 | 53 | 17300 |
14000 |
1.38 |
| H105 | 105 | — | — | 100 | 105 | 34700 |
34800 |
1.53 |
| D77 | 31 | 46 | — | 91 | 28 | 21200 |
15100 |
1.44 |
| T103 | 31 | 46 | 26 | 83 | 47 | 28000 |
20900 |
1.51 |
| TEG111 | — | 111 | — | 0 | 0 | 27600 |
15600 |
1.36 |
The molecular recognition ability of the glycopolymers to concanavalin A (ConA) was determined via fluorescence quenching measurements. The fluorescence intensity change ΔF (F0 – Fn) with changing sugar concentration was measured and analyzed using the Langmuir adsorption isothermal model (ESI†). The obtained binding constants (Ka) are shown in Fig. 3. H-series glycopolymers with mannose were measured. The Ka values of H27, H54 and H105 were 7.5 × 105, 9.0 × 105 and 3.5 × 106 (M−1), respectively. In the case of H-series glycopolymers, the Ka values were on the order of H105 ≫ H54, H27 ≫ monomeric mannose. The Ka values of all H-series glycopolymers were amplified by multivalency and strongly related to the polymer chain length. While the Ka values of H54 and H27 did not show much difference, that of H105 was much larger than those of H54 and H27, suggesting that the binding mode of H105 is different to those of H27–H54. The glycopolymers amplify the sugar–protein interaction by increasing the binding possibility and multivalent binding. The large difference of Ka suggests the bivalent binding of H105 to ConA. The results suggest that the polymer length of H105 is longer than the distance of two sugar-binding sites of 6.5 nm.
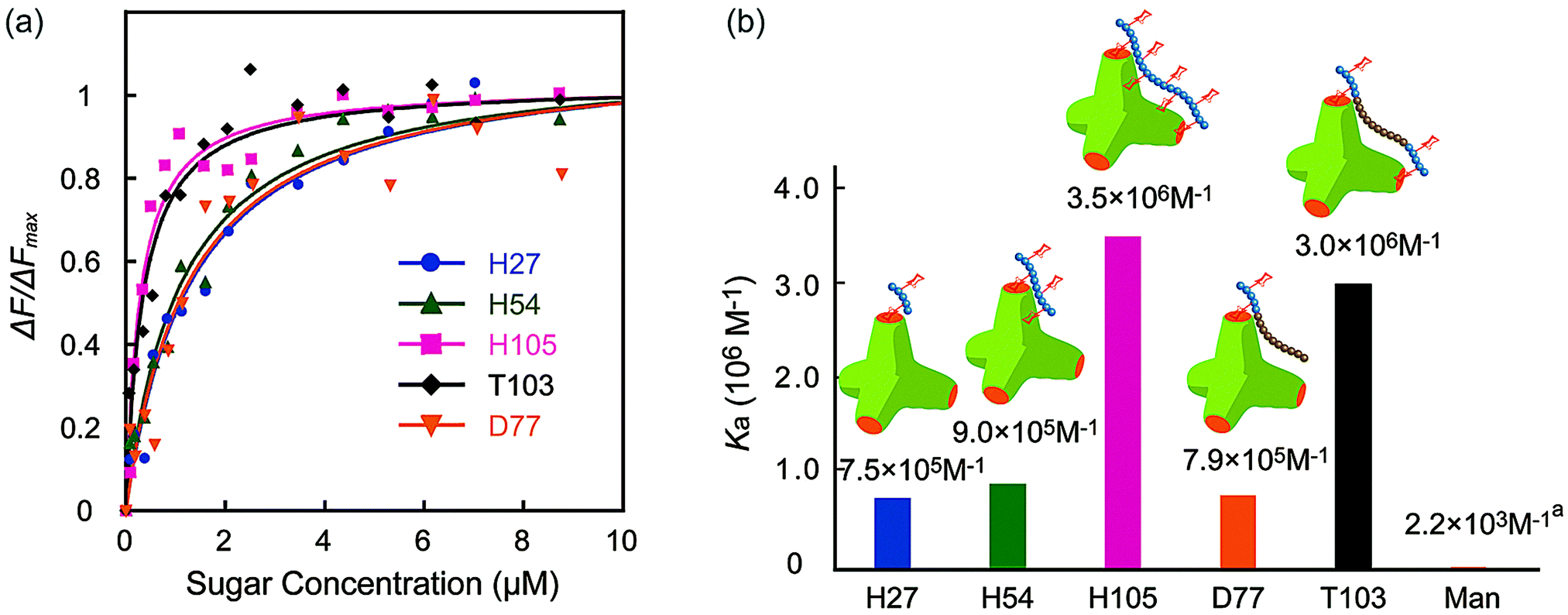 | ||
| Fig. 3 (a) Quenching of fluorescence intensities (FITC-ConA) by the addition of glycopolymers (H27, H54, H105, D77, and T103), and (b) the binding constants of glycopolymers. (In (a) the Ka of mannose is the literature value.21 | ||
Molecular recognition of other polymers was also studied. The Ka values of T103 and D77 were 3.0 × 106 and 7.9 × 105 (M−1), respectively. The Ka value of T103 is comparable to that of H105, and that of D77 is comparable to those of H27 and H54. Taking into consideration all polymers, the order of Ka was H105, T103 ≫ H54, D77, H27 ≫ mannose. The strong molecular recognition of H105 and T103 was also confirmed using a hemagglutination inhibition assay (ESI†).
Interestingly, the tri-block polymer T103 showed almost the same Ka value as that of H105, even though the sugar density in this polymer was almost half that of H105. The molecular length of the two polymers H105 and T103 for the 100-mer was assumed to be longer than the distance of the sugar binding sites. Actually, the tri-block polymer T103 attained multivalent interactions to ConA. Taking into consideration the sugar density of the polymer, the protein recognition efficiency of T103 was much higher than that of H105.
The fluorescence quenching experiments were also performed with FITC-peanut agglutinin (PNA; galactose recognition protein) by the addition of T103 (Fig. 4a). The fluorescence change (ΔF) was not observed. The similar experiment was conducted with FITC-ConA by the addition of TEG111, which also did not induce the fluorescence change (Fig. S9-2, ESI†). The results indicated that the specific interaction of FITC-ConA with the mannose ligand contributed to the fluorescence quenching.
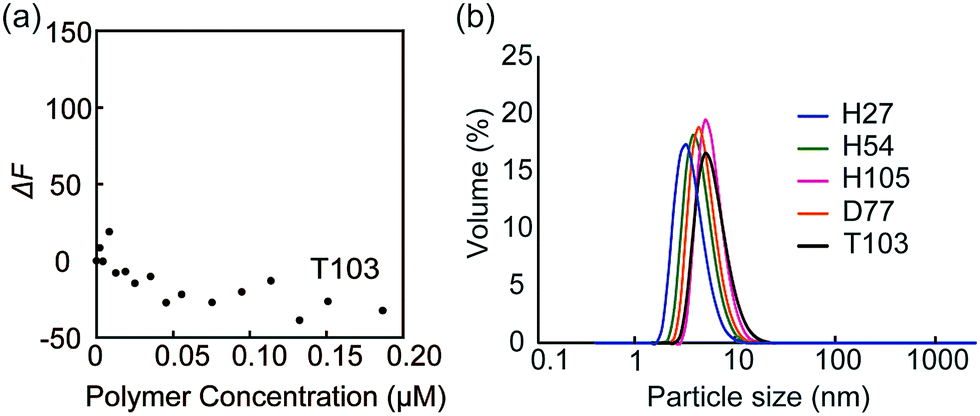 | ||
| Fig. 4 (a) Fluorescence intensity changes of FITC-PNA by the addition of glycopolymer T103, and (b) DLS data of the glycopolymer in PBS buffer. | ||
Dynamic light scattering (DLS) of the glycopolymers was measured to estimate the polymer size in solution (Fig. 4b). The DLS results showed that the sizes of all polymers were less than 10 nm, indicating that the polymers exist in the monomeric state. In the above results shown in Fig. 3, the multivalent interaction of glycopolymers occurs through one polymer chain and does not involve the molecular assembly of polymer-like micelles. That is, the molecular recognition of the glycopolymer can be controlled only by the design of one polymer.
The molecular length of the glycopolymers was determined via a theoretical approach. The monomer size of the glycopolymer was estimated by molecular modeling, and the polymer size was estimated by a conventional Monte Carlo equation.23,24 The calculated molecular lengths of the glycopolymers of H27, H54, H105, D77 and T103 in solution were determined to be 3.7, 5.5, 8.0, 6.7 and 8.0 nm, respectively (ESI†). Since the distance of the sugar binding sites in ConA is 6.5 nm,18 a glycopolymer that exceeds 6.5 nm having a sugar block at both the ends exhibits strong binding due to the two-site binding mode, which is indicated by a marked increase in Ka. The molecular length of T103 was calculated to be more than 6.5 nm, which supports the result of strong binding to ConA. T103 displays a mannose for two-site binding. Although the molecular length of D77 is 6.7 nm, the sugar block (31-mer) was ca. 3.8 nm, which is less than the distance between sugar binding sites, resulting in the weak binding.
Our results of the tri-block copolymer and glycopolymer suggest that living radical polymerization is a suitable method to control ligand platform features, and that the block polymer enlarged the diversity of the glycopolymer and bio-functional polymer design. In the structure of the glycopolymer, the multivalent glycopolymer was found to be a key feature of strong binding and the localization of the sugar cluster in the polymer ensures the multivalent effect of longer glycopolymers. In this investigation, we inserted poly(TEG-MA) into the middle of the polymer because of its biologically inert properties. The poly(TEG-MA) domain can change other polymers with other functional groups like electrostatic, photoactive and electroactive groups, yielding higher functionalized polymers while maintaining the molecular recognition ability.
The sugar arrangement in the glycopolymers was controlled by RAFT living radical polymerization. The molecular recognition ability of glycopolymers was determined by the sugar arrangement to lectin, but not by sugar density in the polymer. The Ka value of the glycopolymer of around 100-mer was 5 times larger than those of the shorter glycopolymers. Both the homopolymer and the tri-block copolymer also showed strong molecular recognition ability. When the theoretical distance between the molecular chain terminals was close to the distance between the binding sites, multipoint binding was achieved. Multipoint binding with lectin was achieved by controlling the spatial arrangement of the ligand in the tri-block glycopolymer composed of blocks of different physical properties without introducing a sugar chain between the binding sites. Control measurements revealed that the glycopolymers used in this study show specific binding to ConA with a single mannose. These results show that the molecular recognition ability with lectin can be designed and controlled by regulating the spatial arrangement of the ligand in the glycopolymer, and the results provide a guide that aims at realizing the de novo design of synthetic polymers that enable advanced and precise functions.
This work was supported by a Grant-in-Aid for Scientific Research (B) (Grant No. JP15H03818) and a Grant-in-Aid for Challenging Exploratory Research (JP16K14007).
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Huang, S. Boyken and D. Baker, Nature, 2016, 537, 320–327 CrossRef CAS PubMed.
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS PubMed.
- W. A. Braunecker and K. Matyjaszewski, Polym. Sci., 2007, 32, 93–146 CAS.
- M. Ouchi and M. Sawamoto, Macromolecules, 2017, 50, 2603–2614 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- M. R. Hill, R. N. Carmean and B. S. Sumerlin, Macromolecules, 2015, 48, 5459–5469 CrossRef CAS.
- L. Martin, G. Gody and S. Perrier, Polym. Chem., 2015, 6, 4875–4886 RSC.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 1–10 RSC.
- N. G. Engelis, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Nat. Chem., 2016, 9, 171–178 CrossRef PubMed.
- Y. Miura, Y. Hoshino and H. Seto, Chem. Rev., 2016, 116, 1673–1692 CrossRef CAS PubMed.
- P. Bojarová and V. Křen, Biomater. Sci., 2016, 4, 1142–1160 RSC.
- J. E. Gestwicki, C. W. Cairo, L. E. Strong, K. A. Oetjen and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 14922–14933 CrossRef CAS PubMed.
- M. Waldmann, R. Jirmann, K. Hoelscher, M. Wienke, F. C. Niemeyer, D. Rehders and B. Meyer, J. Am. Chem. Soc., 2014, 136, 783–788 CrossRef CAS PubMed.
- T. Hasegawa and T. Sasaki, Chem. Commun., 2003, 978–979 RSC.
- K. Matsuura, M. Hibino, Y. Yamada and K. Kobayashi, J. Am. Chem. Soc., 2001, 123, 357–358 CrossRef CAS PubMed.
- J. Greer, H. W. Kaufman and A. J. Kalb, J. Mol. Biol., 1970, 48, 365–366 CrossRef CAS PubMed.
- Z. Derewenda, J. Yariv, J. R. Helliwell, A. J. Kalb, E. J. Dodson, M. Z. Papiz, T. Wan and J. Campbell, EMBO J., 1989, 8, 2189–2193 CAS.
- T. Hasegawa, S. Kondoh, K. Matsuura and K. Kobayashi, Macromolecules, 1999, 32, 6595–6603 CrossRef CAS.
- Y. Miura, D. Koketsu and K. Kobayashi, Polym. Adv. Technol., 2007, 18, 647–651 CrossRef CAS.
- T. K. Dam, R. Roy, S. K. Das, S. Oscarson and C. F. Brewer, J. Biochem., 2000, 275, 14223–14230 CAS.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
- C. M. Guttman and E. A. DiMarzio, Macromolecules, 1982, 15, 525–531 CrossRef CAS.
- I. Teraoka, Polymer Solutions: An Introduction to Physical Properties, John Wiley & Sons, Hoboken, 2002 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc07107h |
| This journal is © The Royal Society of Chemistry 2018 |