
In trans hydrolysis of carrier protein-bound acyl intermediates by CitA during citrinin biosynthesis†
Philip A.
Storm
 and
Craig A.
Townsend
*
and
Craig A.
Townsend
*
Department of Chemistry, Johns Hopkins University, 3400 N Charles St, Baltimore, MD 21218, USA. E-mail: ctownsend@jhu.edu
First published on 30th November 2017
Abstract
Polyketide synthases (PKSs) have several known editing mechanisms to ensure that non-productive intermediates are removed from the acyl carrier protein (ACP). We demonstrate that CitA, a putative hydrolase in the citrinin biosynthetic gene cluster, removes ACP-bound acyl intermediates. We propose that it serves an editing role in trans.
Polyketide synthases (PKSs) are multi-domain biosynthetic enzymes that catalyze the formation of complex carbon scaffolds through repeated rounds of extension and modification of simple acyl substrates.1–3 Intermediates are covalently tethered to the PKS by thioester linkage to a post-translationally installed phosphopantetheine arm of the acyl carrier protein (ACP) and delivered to the active sites of other domains for programmed chemistry. Mature intermediates are released from the ACP through a number of mechanisms including hydrolysis, cyclization, reduction, or transfer to a downstream carrier protein. The reactive nature of many PKS intermediates, however, occasionally results in the presence of acyl-holo-ACP species that are not on-path to the programmed PKS product and must be removed for productive chemistry to resume. This reactivity is fundamentally problematic for non-reduced polyketide intermediates that juxtapose nucleophilic and electrophilic moieties susceptible to intramolecular reactions that derail the acyl intermediate from the programmed path, for example the formation of the SEK4 and SEK4b derailment products.4,5
Ensuring that the genetic and metabolic investment in such large biosynthetic machinery is not perpetually waylaid, several different strategies to remove unproductive acyl intermediates have been employed across multiple types of PKSs. Many Type I PKSs have a thioesterase (TE) domain at their C-terminus commonly associated with product release and some also have hydrolytic activity towards other acyl-holo-ACP species.6 In some cases, however, this hydrolytic activity has been maintained by other proteins acting in trans. Fungal non-reducing PKSs (NR-PKSs) lacking a C-terminal TE domain can have a separate metallo-β-lactamase type TE in the same gene cluster that hydrolyzes the mature ACP-bound intermediate as in atrochrysone carboxylic acid biosynthesis.7
In bacterial trans-AT PKSs, where extender units are loaded onto the ACP by a discrete acyltransferase, the AT homolog PedC was recently shown to serve as a stand-alone acyl hydrolase (AH) capable of liberating several acyl species from the ACP in pederin biosynthesis.8 The presence of in trans hydrolysis mechanisms across disparate PKSs suggests that this activity may represent a general strategy to maintain PKS efficiency in the absence of a cis-TE (Fig. 1).
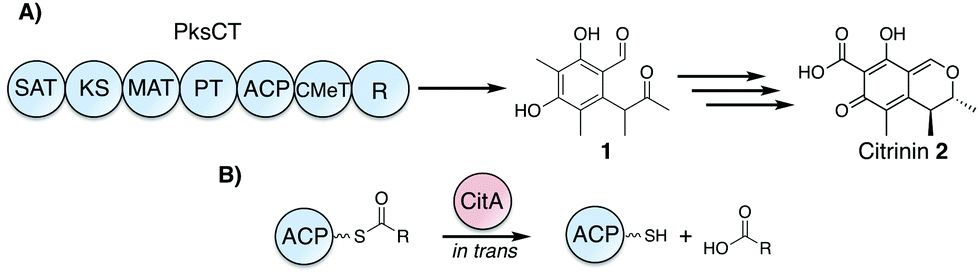 | ||
| Fig. 1 Citrinin biosynthesis and proposed role of CitA as an in trans hydrolase. (A) PksCT produces the pentaketide aldehyde 1, which is later transformed to citrinin 2 by several redox steps and cyclization to the dihydroisochromene core. (B) CitA is proposed to catalyze the hydrolysis of ACP-bound acyl intermediates to free the ACP from stalled intermediates. (SAT: starter unit-ACP transacylase, KS: ketosynthase, MAT: malonyl-CoA:ACP transacylase, PT: product template, ACP: acyl carrier protein, CMeT: C-methyltransferase, R: reductase). | ||
While many fungal NR-PKSs contain a C-terminal TE that may have an editing role, the recently categorized Group VII PKSs terminate instead with a reductase (R) domain that catalyzes NADPH-dependent release of the mature thioester intermediate as an aldehyde.9 Whether R domains are capable of carrying out an analogous editing function is unclear, but such a function would presumably involve the expenditure of NADPH and be energetically expensive for the producing organism relative to hydrolysis. Additional work has also identified the presence of a putative α/β-hydrolase-encoding gene adjacent to Group VII PKSs in a number of biosynthetic gene clusters, and co-expression of the PKS and hydrolase gives greater titers of the post-PKS product compared to expression of the PKS alone.10,11 Several roles for such putative hydrolases have been proposed, and uncertainty was exacerbated by the initial misannotation of these genes.12 While their exact function is not resolved, in trans editing of acyl intermediates is a possible role for these accessory proteins and one consistent with the in vivo results.
We sought to identify the activity of CitA (GenBank: BAE95339), the putative hydrolase adjacent to PksCT, the Group VII NR-PKS in the M. purpureus gene cluster responsible for citrinin biosynthesis.13,14 Previously, CitA was re-annotated from an oxidoreductase to a hydrolase due to an incorrect initial assignment of the start codon, and deletion of CitA in the citrinin-producer Monascus ruber greatly decreased, but did not eliminate the post-PKS aldehyde 1.10 Additionally, co-expression of both PksCT and CitA in the heterologous host Aspergillus oryzae gave significantly higher titers than the PKS alone. Based only on these observations, however, the role of CitA could not be established definitively. Using a previously reported domain-deconstructed system, we added CitA to in vitro reconstituted PksCT to assess its effect on the characterized product profiles from specific domain combinations.15 Addition of CitA to all combinations of PksCT domains resulted in a concentration-dependent decrease in derailment products triketides 3 and 4, tetraketide 5, and pentaketide 6, as well as the on-path product aldehyde 1 (Fig. 2). No new products were found as might be expected if CitA catalyzed a modification of PKS-bound intermediates or the final product 1. Unlike reported results of in vivo coexpression of PksCT with CitA, we did not observe any increase in 1 at any concentration of CitA. We suspect that synthetic inefficiencies from dissection may explain the lack of increase in 1in vitro. The in vivo use of intact PksCT allows for intramolecular interaction between the ACP and other PksCT domains, though we could not obtain soluble intact PksCT from heterologous expression in E. coli or Saccharomyces cerevisiae to test this hypothesis.
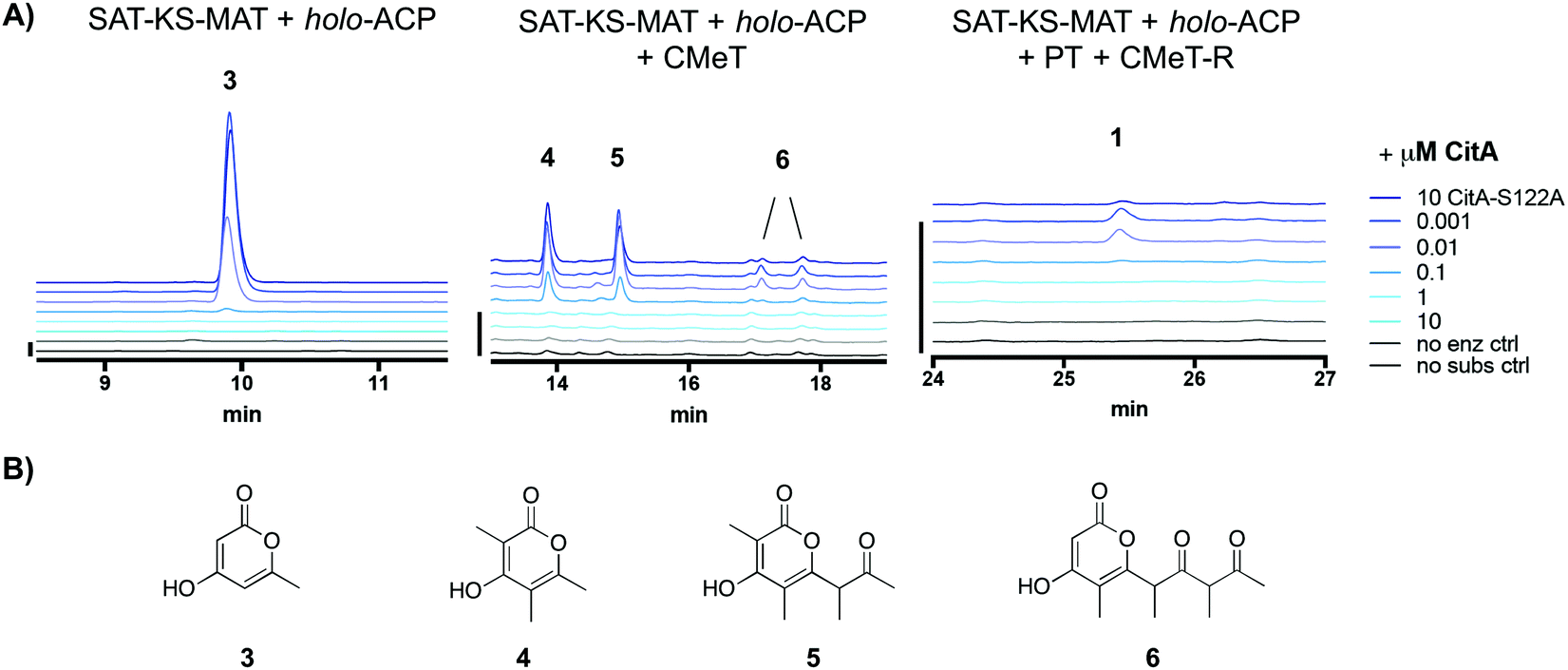 | ||
| Fig. 2 CitA reduces production of derailment products and the on-path aldehyde 1 in PksCT reconstitution reactions. (A) Product profiles of various PksCT domain combinations with increasing concentrations of CitA, performed as previously described in ref. 13, with absorbance at 280 nm. The minimal PKS reaction produces only the triketide 3. The minimal PKS with CMeT produces tri-, tetra-, and pentaketides 4, 5, and 6. The fully reconstituted PksCT produces small amounts of 1. Scale bars at the left of each set of traces indicate 10 mAU. (B) Structures of the spontaneously released pyrone derailment products. | ||
Since CitA only appeared to decrease product yields, we speculated that it might be hydrolyzing one or more intermediates, analogous to a recent observation of in trans acyl-holo-ACP hydrolysis in pederin biosynthesis.8 We identified a GxSxG motif common to α/β-hydrolases that was conserved in several putative CitA-like hydrolases adjacent to Group VII NR-PKSs, including AfoE, PkdA, PkeA, and PkhA (Fig. S1, ESI†).9,16,17 We also generated a homology model of CitA using the CPHmodels 3.2 server, which identified the yeast serine hydrolase FSH1/YHR049W as the closest homolog of known structure (PDB: 1YCD).18,19 The homology model suggested a Ser122–His235–Asp207 catalytic triad, consistent with recent in vivo observations of the CitA homolog MppD in azaphilone biosynthesis in M. purpureus.11 To test CitA hydrolysis against the simplest of acyl-ACP species, PksCT apo-ACP (ACPCT) monodomain was activated by the promiscuous phosphopantetheinyltransferase Sfp with [1-14C]-acetyl-CoA to give the radiolabeled acetyl-holo-ACPCT and incubated with CitA or CitA-S122A. Following separation of the reaction products by SDS-PAGE, we observed that the acetyl radiolabel was lost from ACPCT in a CitA-dependent fashion, but CitA-S122A was inert (Fig. 3A). A radiolabeled band consistent with the larger CitA was not detected in the gel, suggesting that CitA does not retain the acetyl species for transfer to a downstream acceptor but rapidly hydrolyzes it to free acetate.
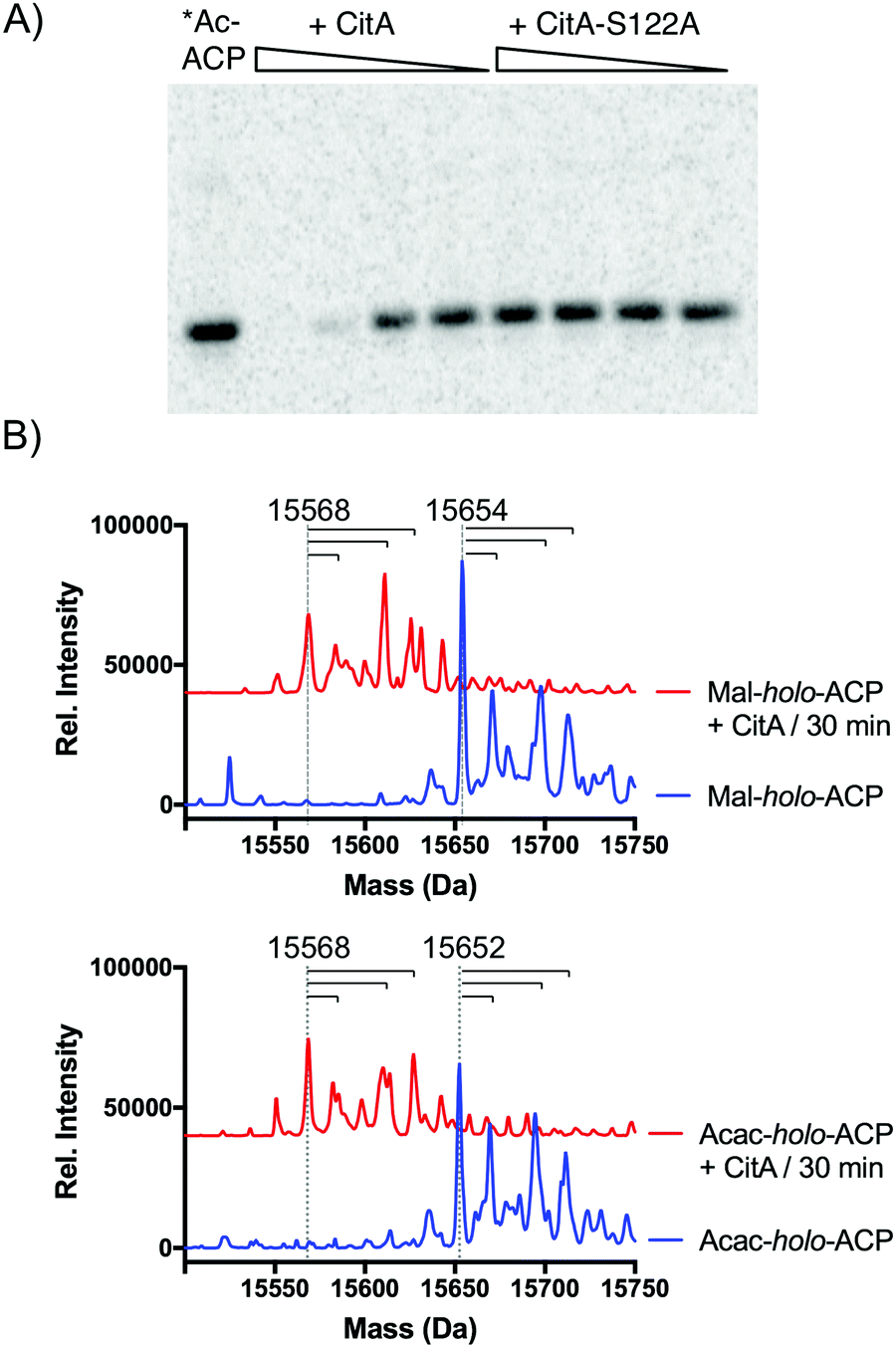 | ||
| Fig. 3 CitA hydrolyzes ACP-bound acyl intermediates. (A) Incubation of [1-14C]-acetyl-holo-ACPCT with CitA results in loss of the radiolabel in a concentration-dependent fashion. Mutation of the putative nucelophile Ser122 to Ala rendered CitA inactive. No transfer of the radiolabel to CitA was observed. (B) Malonyl-holo-ACPCT (15654 Da) and acetoacetyl-holo-ACPCT (15652 Da) are also hydrolyzed to free holo-ACPCT (15568 Da) by CitA. The dashed vertical lines mark the labeled masses and the horizontal lines correspond to +16, +42, and +58 Da masses consistent with oxidation, acetylation, and oxidation plus acetylation. These modifications are also observed in the apo-ACP. Red traces are vertically offset for clarity. | ||
Previous examples of editing TEs or hydrolases in PKS biosynthesis have been shown to have varying degrees of substrate promiscuity towards both on-path and likely off-path intermediates.6,8 Many of the acyl-ACPCT intermediates en route to 1 are not accessible due to their inherent reactivity, preventing analysis of late-stage tri-, tetra-, or pentaketide intermediates. However, we tested the hydrolytic activity of CitA against malonyl-holo-ACPCT and acetoacetyl-holo-ACPCT – acyl species bound to ACPCT at early stages of the biosynthetic cycle. The mass of the acyl-ACPs was determined by UPLC-ESI-MS before and after incubation with CitA (Fig. 3B and Fig S4–S8, ESI†). apo-ACPCT was observed as a quartet of masses consistent with oxidation, acetylation, and both modifications (Fig. S4, ESI†) that we also observed throughout these experiments.20 Loss of mass consistent with acyl hydrolysis was observed for both malonyl and acetoacetyl substrates. Competition experiments combining malonyl- and acetoacetyl-holo-ACPCT did not show a significant substrate preference by CitA (Fig. S9, ESI†). Attempts to obtain Michaelis–Menten values to compare these acyl species using the N-acetylcysteamine (SNAC) or CoA thioesters as substrate mimics were unsuccessful due to an inability to saturate CitA activity. The weak activity against small molecule substrates suggested that the ACP is an essential component of CitA substrates, focusing CitA activity towards PKS-bound intermediates and limiting indiscriminant hydrolysis of acyl-CoA species.
Several investigations into the interactions of ACP domains with PKS or fatty acid synthase client domains have identified conserved basic residues that form contacts with the phosphodiester moiety of the phosphopantetheine arm or conserved acidic residues on the acyl-holo-ACP, including product template, ketosynthase, and acyltransferase domains.21–25 Using our homology model in conjunction with sequence alignments of CitA to related hydrolases, we identified two basic residues – Arg36 and Arg236 – near the entrance of the CitA active site that could be involved in the CitA:ACPCT binding interface (Fig. 4A and Fig. S2, S3 (ESI†) for characterization). Both are predicted to be within ∼20 Å of Ser122, approximately the length of the extended phosphopantetheine arm. CitA-R36A and CitA-R236A mutants were generated and incubated with [1-14C]-acetyl-holo-ACPCT in a time-course assay and separated by SDS-PAGE (Fig. 4B). CitA-R236A was indistinguishable from the wild type and both completely hydrolyzed the acetyl radiolabel. In contrast, CitA-R36A had minimal hydrolytic activity suggesting that Arg36 has an essential role in mediating the in trans interaction between acyl-holo-ACPCT and CitA. The conserved nature of this basic residue among CitA homologs in other fungal NR-PKS biosynthetic gene clusters reinforces the conclusion that CitA-like hydrolases preferentially act upon PKS-bound acyl intermediates.
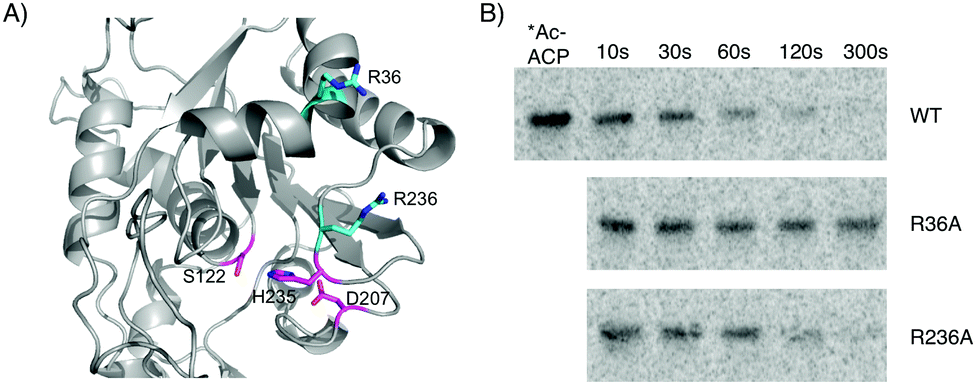 | ||
| Fig. 4 Interaction of CitA with acyl-holo-ACPCT is dependent on Arg36. (A) Homology modeling of CitA shows two conserved Arg residues (cyan) at the opening of the active site and approximately 20 Å from Ser122, the nucleophile in the conserved catalytic triad (magenta). (B) Time-course incubation of CitA (1 μM) with [1-14C]-acetyl-holo-ACPCT (10 μM) shows significantly reduced hydrolysis by the R36A mutant compared to wild type. The R236A mutant had minimal effect on hydrolysis. | ||
In conclusion, our results expand the scope of in trans editing of acyl-ACP intermediates in fungi, and highlight it as a generalized strategy to ensure efficient polyketide biosynthesis, especially when the PKS lacks other self-editing mechanisms. We show that several carrier protein-bound acyl intermediates are removed by CitA and identify conserved features that are essential to CitA:ACP binding and hydrolysis. While acetyl-holo-ACPCT is an on-path intermediate to initiate citrinin biosynthesis, hydrolysis of acetyl-ACPCT by CitA prevents stalling of PksCT in the event of spontaneous, non-productive decarboxylation of malonyl-holo-ACPCT during extension of longer chain-length intermediates. Additionally, hydrolytic activity towards malonyl or acetoacetyl intermediates may be necessary when other client domains like the ketosynthase, C-methyltransferase, or reductase have lost their bound co-substrates or have been otherwise inactivated. It remains unclear how PksCT and CitA collaborate in producing organisms, as the in vitro data do not replicate the increased yield seen in vivo. Acyl-holo-ACPCT species may be less exposed in intact PksCT than in our dissected system due to faster intramolecular shuttling of ACP-bound intermediates, favoring binding of ACPCT to other PksCT client domains relative to intermolecular association with CitA. When PksCT is dissected, all ACPCT interactions occur in trans, with hydrolysis by CitA expectedly competing against polyketide synthesis. Further study of relative expression, catalytic rates, precursor availability, and other factors affecting efficiency may uncover the optimal balance between editing and productive biosynthesis. As a significant number of fungal and bacterial PKSs have been found to adopt in trans editing mechanisms, future work to engineer these polyketide pathways, or any others lacking metabolically optimized editing, may benefit by including an in trans hydrolase, perhaps under distinct and tunable control.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC.
- J. M. Crawford and C. A. Townsend, Nat. Rev. Microbiol., 2010, 8, 879–889 CrossRef CAS PubMed.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS PubMed.
- A. G. Newman, A. L. Vagstad, P. A. Storm and C. A. Townsend, J. Am. Chem. Soc., 2014, 136, 7348–7362 CrossRef CAS PubMed.
- H. Fu, D. A. Hopwood and C. Khosla, Chem. Biol., 1994, 1, 205–210 CrossRef CAS PubMed.
- A. L. Vagstad, S. B. Bumpus, K. Belecki, N. L. Kelleher and C. A. Townsend, J. Am. Chem. Soc., 2012, 134, 6865–6877 CrossRef CAS PubMed.
- T. Awakawa, K. Yokota, N. Funa, F. Doi, N. Mori, H. Watanabe and S. Horinouchi, Chem. Biol., 2009, 16, 613–623 CrossRef CAS PubMed.
- M. Jenner, J. P. Afonso, C. Kohlhaas, P. Karbaum, S. Frank, J. Piel and N. J. Oldham, Chem. Commun., 2016, 52, 5262–5265 RSC.
- M. Ahuja, Y. M. Chiang, S. L. Chang, M. B. Praseuth, R. Entwistle, J. F. Sanchez, H. C. Lo, H. H. Yeh, B. R. Oakley and C. C. Wang, J. Am. Chem. Soc., 2012, 134, 8212–8221 CrossRef CAS PubMed.
- Y. He and R. J. Cox, Chem. Sci., 2016, 7, 2119–2127 RSC.
- B. Balakrishnan, R. Chandran, S. H. Park and H. J. Kwon, J. Microbiol. Biotechnol., 2015, 25, 1648–1652 CrossRef CAS PubMed.
- J. Davison, A. al Fahad, M. Cai, Z. Song, S. Y. Yehia, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7642–7647 CrossRef CAS PubMed.
- T. Shimizu, H. Kinoshita, S. Ishihara, K. Sakai, S. Nagai and T. Nihira, Appl. Environ. Microbiol., 2005, 71, 3453–3457 CrossRef CAS PubMed.
- T. Shimizu, H. Kinoshita and T. Nihira, Appl. Environ. Microbiol., 2007, 73, 5097–5103 CrossRef CAS PubMed.
- P. A. Storm, D. A. Herbst, T. Maier and C. A. Townsend, Cell Chem. Biol., 2017, 24, 316–325 CrossRef CAS PubMed.
- Y. M. Chiang, E. Szewczyk, A. D. Davidson, N. Keller, B. R. Oakley and C. C. Wang, J. Am. Chem. Soc., 2009, 131, 2965–2970 CrossRef CAS PubMed.
- S. Brenner, Nature, 1988, 334, 528–530 CrossRef CAS PubMed.
- M. Nielsen, C. Lundegaard, O. Lund and T. N. Petersen, Nucleic Acids Res., 2010, 38, W576–581 CrossRef CAS PubMed.
- S. Quevillon-Cheruel, N. Leulliot, M. Graille, N. Hervouet, F. Coste, H. Benedetti, C. Zelwer, J. Janin and H. Van Tilbeurgh, Protein Sci., 2005, 14, 1350–1356 CrossRef CAS PubMed.
- J. Zhang, R. Sprung, J. Pei, X. Tan, S. Kim, H. Zhu, C. F. Liu, N. V. Grishin and Y. Zhao, Mol. Cell. Proteomics, 2009, 8, 215–225 CAS.
- K. J. Weissman and R. Muller, ChemBioChem, 2008, 9, 826–848 CrossRef CAS PubMed.
- J. F. Barajas, G. Shakya, G. Moreno, H. Rivera, Jr., D. R. Jackson, C. L. Topper, A. L. Vagstad, J. J. La Clair, C. A. Townsend, M. D. Burkart and S. C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E4142–E4148 CrossRef CAS PubMed.
- A. Miyanaga, S. Iwasawa, Y. Shinohara, F. Kudo and T. Eguchi, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1802–1807 CrossRef CAS PubMed.
- C. Nguyen, R. W. Haushalter, D. J. Lee, P. R. Markwick, J. Bruegger, G. Caldara-Festin, K. Finzel, D. R. Jackson, F. Ishikawa, B. O'Dowd, J. A. McCammon, S. J. Opella, S. C. Tsai and M. D. Burkart, Nature, 2014, 505, 427–431 CrossRef CAS PubMed.
- J. Bruegger, R. W. Haushalter, A. L. Vagstad, G. Shakya, N. Mih, C. A. Townsend, M. D. Burkart and S. C. Tsai, Chem. Biol., 2013, 20, 1135–1146 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc07079a |
| This journal is © The Royal Society of Chemistry 2018 |