
Intra-hydrogel culture prevents transformation of mesenchymal stem cells induced by monolayer expansion

Abstract
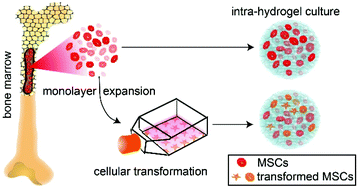
In this study, we report that the intra-hydrogel culture system mitigates the transformation of mesenchymal stem cells (MSCs) induced by two-dimensional (2D) expansion. MSCs expanded in monolayer culture prior to encapsulation in collagen hydrogels (group eMSCs-CH) featured impaired stemness in chondrogenesis, comparing with the freshly isolated bone marrow mononuclear cells seeded directly in collagen hydrogels (group fMSCs-CH). The molecular mechanism of the in vitro expansion-triggered damage to MSCs was detected through genome-wide microarray analysis. Results indicated that pathways such as proteoglycans in cancer and pathways in cancer expansion were highly enriched in eMSCs-CH. And multiple up-regulated oncoma-associated genes were verified in eMSCs-CH compared with fMSCs-CH, indicating that expansion in vitro triggered cellular transformation was associated with signaling pathways related to tumorigenicity. Besides, focal adhesion (FA) and mitogen-activated protein kinase (MAPK) signaling pathways were also involved in in vitro expansion, indicating restructuring of the cell architecture. Thus, monolayer expansion in vitro may contribute to vulnerability of MSCs through the regulation of FA and MAPK. This study indicates that intra-hydrogel culture can mitigate the monolayer expansion induced transformation of MSCs and maintain the uniformity of the stem cells, which is a viable in vitro culture system for stem cell therapy.
 Please wait while we load your content...
Please wait while we load your content...

