
Protein-engineered hydrogels enhance the survival of induced pluripotent stem cell-derived endothelial cells for treatment of peripheral arterial disease†
 ab
and
Ngan F.
Huang
*bcd
ab
and
Ngan F.
Huang
*bcd
Abstract
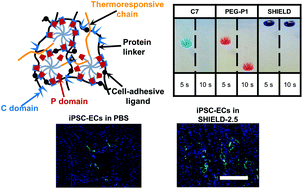
A key feature of peripheral arterial disease (PAD) is damage to endothelial cells (ECs), resulting in lower limb pain and restricted blood flow. Recent preclinical studies demonstrate that the transplantation of ECs via direct injection into the affected limb can result in significantly improved blood circulation. Unfortunately, the clinical application of this therapy has been limited by low cell viability and poor cell function. To address these limitations we have developed an injectable, recombinant hydrogel, termed SHIELD (Shear-thinning Hydrogel for Injectable Encapsulation and Long-term Delivery) for cell transplantation. SHIELD provides mechanical protection from cell membrane damage during syringe flow. Additionally, secondary in situ crosslinking provides a reinforcing network to improve cell retention, thereby augmenting the therapeutic benefit of cell therapy. In this study, we demonstrate the improved acute viability of human induced pluripotent stem cell-derived endothelial cells (iPSC-ECs) following syringe injection delivery in SHIELD, compared to saline. Using a murine hind limb ischemia model of PAD, we demonstrate enhanced iPSC-EC retention in vivo and improved neovascularization of the ischemic limb based on arteriogenesis following transplantation of iPSC-ECs delivered in SHIELD.
 Please wait while we load your content...
Please wait while we load your content...

