
A new photoelectrochemical biosensor for ultrasensitive determination of nucleic acids based on a three-stage cascade signal amplification strategy†
Erhu
Xiong‡
,
Xiaoxia
Yan‡
,
Xiaohua
Zhang
 *,
Yanmei
Li
,
Ruiying
Yang
,
Leixia
Meng
and
Jinhua
Chen
*
*,
Yanmei
Li
,
Ruiying
Yang
,
Leixia
Meng
and
Jinhua
Chen
*
State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, P. R. China. E-mail: chenjinhua@hnu.edu.cn; mickyxie@hnu.edu.cn
First published on 12th May 2018
Abstract
The sensitive and specific determination of nucleic acids is very important in clinical diagnosis and biological studies. In this work, an ultrasensitive photoelectrochemical (PEC) biosensor has been developed for DNA detection based on a “signal-on” sensing strategy and a three-stage cascade signal amplification method (catalytic hairpin assembly (CHA), hybridization chain reaction (HCR) and alkaline phosphatase (ALP)-triggered in situ generation of ascorbic acid (AA)). Here, CHA hairpin 1 (CHA-HP1) is opened by the target DNA (T-DNA) owing to the hybridization between T-DNA and CHA-HP1, and then the opened CHA-HP1 hybridizes with CHA hairpin 2 (CHA-HP2) to displace the T-DNA, generating a CHA-HP1/CHA-HP2 complex. The displaced T-DNA triggers the next cycle of CHA, resulting in the generation of numerous CHA-HP1/CHA-HP2 complexes. Subsequently, one end of the CHA-HP1/CHA-HP2 complex hybridizes with the capture DNA immobilized on the indium tin oxide/TiO2/CdS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn electrode. After the introduction of dual-biotin labeled HCR hairpin 1 (HCR-HP1-Bio) and dual-biotin labeled HCR hairpin 2 (HCR-HP2-Bio), the other end of the CHA-HP1/CHA-HP2 complex opens HCR-HP1-Bio. The opened HCR-HP1-Bio triggers the HCR reaction between HCR-HP1-Bio and HCR-HP2-Bio, leading to the formation of long nicked duplex DNA structures. The dual-biotin modified HCR-hairpins can anchor more streptavidin–ALP to catalyze 2-phospho-L-ascorbic acid trisodium salt to yield more AA, leading to a larger PEC response. The proposed PEC biosensor shows superior analytical performance for T-DNA detection with a linear response ranging from 0.1 fM to 100 pM and a detection limit of 0.052 fM, and may provide a powerful biosensing platform for bioanalysis and early disease diagnosis.
:Mn electrode. After the introduction of dual-biotin labeled HCR hairpin 1 (HCR-HP1-Bio) and dual-biotin labeled HCR hairpin 2 (HCR-HP2-Bio), the other end of the CHA-HP1/CHA-HP2 complex opens HCR-HP1-Bio. The opened HCR-HP1-Bio triggers the HCR reaction between HCR-HP1-Bio and HCR-HP2-Bio, leading to the formation of long nicked duplex DNA structures. The dual-biotin modified HCR-hairpins can anchor more streptavidin–ALP to catalyze 2-phospho-L-ascorbic acid trisodium salt to yield more AA, leading to a larger PEC response. The proposed PEC biosensor shows superior analytical performance for T-DNA detection with a linear response ranging from 0.1 fM to 100 pM and a detection limit of 0.052 fM, and may provide a powerful biosensing platform for bioanalysis and early disease diagnosis.
Introduction
The specific and sensitive determination of nucleic acids is very important in clinical diagnosis, mutation detection, and gene therapy. Therefore, developing sensitive methods for the determination of trace target nucleic acid sequences becomes a significant issue. In recent years, there are many sorts of approaches that have been developed, such as colorimetry,1,2 electrochemistry,3–5 fluorescence,6–9 surface plasmon resonance,10,11 chemiluminescence,12,13 electrochemiluminescence,14,15 and photoelectrochemistry.16–20 Among these approaches, the photoelectrochemical (PEC) method has attracted substantial attention benefiting from its advantages such as low cost, rapid response, low background signals, high sensitivity, simple instrumentation, and easy miniaturization.17,21,22 Moreover, due to the combination of the electrochemical assay and light irradiation, the PEC biosensor has the advantages of both electrochemical biosensors and optical techniques.In comparison with traditional optical and electrochemical methods, the photoelectric conversion efficiency is a crucial parameter for PEC sensitive assay. As we all know, the photoelectric conversion efficiency mainly relies on the photoactive materials immobilized on the electrode surface. TiO2, as an excellent photoelectric material, has been extensively utilized in PEC biosensors because of its high stability, good biocompatibility, low cost, and environmental friendliness.23–26 However, as a semiconductor material with a wide energy band gap (∼3.2 eV), TiO2 mainly absorbs ultraviolet light, which leads to the inadequate utilization of optical energy and may inevitably cause the damage of biomolecules. CdS is a frequently used semiconductor with a narrower energy band gap (∼2.4 eV) and its absorption range can extend to the medium wavelength region.27 On the other hand, CdS has a higher conduction band than TiO2, which is beneficial to the injection of photogenerated electrons from CdS to TiO2. Furthermore, Mn-doping in CdS can produce new midgap centres, which can effectively inhibit electron–hole recombination.28 The co-sensitized structure with cascade band-edge levels is extremely effective for the PEC biosensor to augment the optical absorption efficiency, to prolong the lifetime of charge carriers and to promote the electron transfer.29,30 In addition, as an excellent electron donor, ascorbic acid (AA) is usually used in the PEC biosensor. AA can rapidly consume the photogenerated holes to yield an oxidized product and to inhibit electron–hole recombination, resulting in an enhanced photocurrent signal (Scheme 1a).31
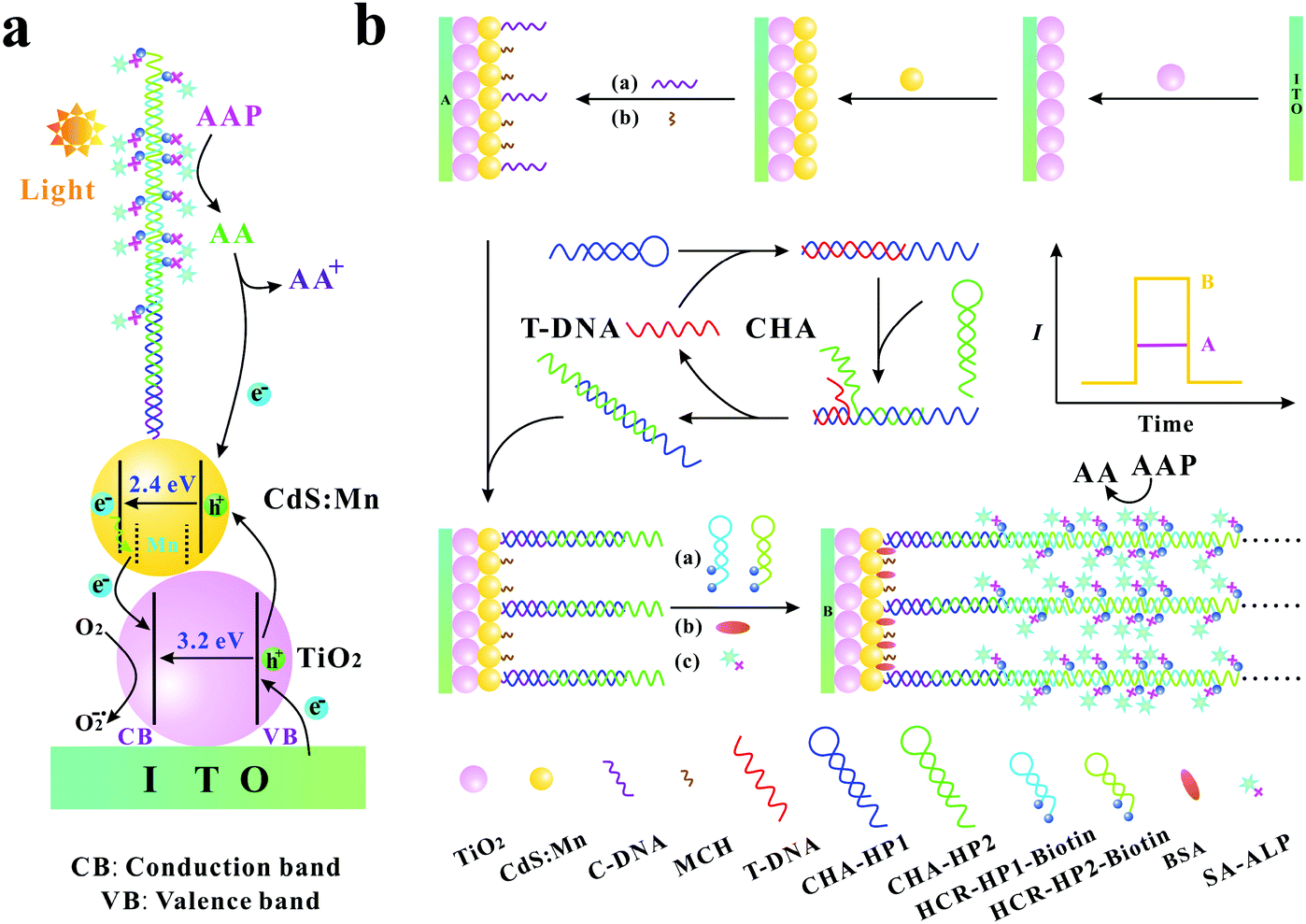 | ||
| Scheme 1 (a) Photogenerated electron–hole transfer mechanism of the biosensing system; (b) schematic illustration of the PEC biosensor for T-DNA detection. | ||
For the analytical performance of the PEC biosensor, the other crucial factor is the effective signal amplification methods. Recently, many kinds of signal amplification methods have been reported, such as hybridization chain reaction (HCR),32–34 ligase chain reaction (LCR),35 polymerase chain reaction (PCR),36 rolling circle amplification (RCA),37,38 catalytic hairpin assembly (CHA),39–41 helicase-dependent amplification (HDA),42,43 strand displacement amplification (SDA)44,45 and so on. Among these signal amplification methods, CHA is an enzyme-free nucleic acid-based signal amplification method that provides a useful means for both transducing and amplifying signals from nucleic acid analytes. In the CHA method, two hairpins cannot hybridize with each other initially, but can catalytically form double-stranded structures in the presence of an initiator, resulting in hundred-fold catalytic amplification.46–49 HCR is another typical and well-known enzyme-free signal amplification method, which is triggered by an initiator and depends on the use of two hairpins to propagate a HCR event.50 The two hairpins in HCR are used as fuel packets to drive the double-stranded DNA propagation.51 The initiator opens one of the two stable hairpins and triggers the HCR event to generate long nicked duplexes, which exhibits awesome potential in the signal amplification of DNA detection.46 Recently, Zang et al. reported that CHA-programmed porphyrin–DNA complexes trigger the chemiluminescence as PEC initiators for the determination of DNA with a low detection limit,52 and Li et al. developed a label-free PEC biosensor for DNA detection via the HCR amplification strategy.18 However, most of these PEC biosensors are just based on one kind of signal amplification strategy (CHA or HCR). Therefore, Ge et al. reported a “signal-off” and two-stage cascade amplification strategy based on CHA and HCR for cancer biomarker detection.53 Nevertheless, it is well known that “signal-off” biosensors are limited by signaling capacity with a maximum of 100% signal suppression.54 Hence, to avoid the shortcoming of the “signal-off” method, it is still of great importance to develop novel “signal-on” PEC biosensors based on the multi-stage signal amplification strategy. The specific advantages of the “signal-on” method, CHA and HCR signal amplification methods, and the important role of AA in the PEC biosensors inspire us to explore the possibility of the development of a new “signal-on” PEC sensing platform with the combination of CHA and HCR signal amplification methods and in situ generation of AA.
In this work, based on the three-stage cascade signal amplification strategy (CHA, HCR and ALP-triggered in situ generation of AA), we developed a novel and ultrasensitive “signal-on” PEC biosensor for the detection of target DNA (T-DNA). Here, human T-cell lymphotropic virus type II DNA (HTLV-II DNA), which has a substantial impact on myelopathy and is also related to urinary tract health, pulmonary symptoms, and incremental mortality,55,56 was selected as the model. As shown in Scheme 1b, CHA hairpin 1 (CHA-HP1) is opened by the T-DNA owing to the hybridization between the complementary sections on the T-DNA and CHA-HP1. Then, the opened CHA-HP1 assembles with CHA hairpin 2 (CHA-HP2) to displace the T-DNA and to generate the CHA-HP1/CHA-HP2 complex. The displaced T-DNA can trigger the next cycle of the CHA process, resulting in the generation of numerous CHA-HP1/CHA-HP2 complexes. Subsequently, one end of the CHA-HP1/CHA-HP2 complex hybridizes with the capture DNA (C-DNA) immobilized on the indium tin oxide (ITO)/TiO2/CdS:Mn electrode. After the introduction of dual-biotin labeled HCR hairpin 1 (HCR-HP1-Bio) and dual-biotin labeled HCR hairpin 2 (HCR-HP2-Bio), the other end of the CHA-HP1/CHA-HP2 complex can open HCR-HP1-Bio. As a trigger, the newly exposed cohesive-end of HCR-HP1-Bio can open HCR-HP2-Bio, and then the opened HCR-HP2-Bio can open HCR-HP1-Bio subsequently. That is, once HCR-HP1-Bio is opened, the HCR reaction between HCR-HP1-Bio and HCR-HP2-Bio occurs, leading to the formation of long nicked duplex DNA structures. Because both the HCR hairpins are labeled with dual-biotin, lots of streptavidin–alkaline phosphatase (SA–ALP) is captured on the electrode surface. Under the enzymatic catalysis of ALP with the substrate 2-phospho-L-ascorbic acid trisodium salt (AAP), AA is yielded in situ as an excellent electron donor, resulting in an enhanced photocurrent response.57,58 Based on the above three-stage cascade signal amplification strategy, the proposed “signal-on” PEC biosensor shows superior analytical performance for the sensitive determination of T-DNA with a detection limit as low as 0.052 fM. As far as we know, there is no report on the “signal-on” PEC biosensors for DNA assay based on the three-stage cascade signal amplification strategy (CHA, HCR and ALP-triggered in situ generation of AA). The developed method may provide a sensitive and powerful biosensing platform for bioanalysis and early disease diagnosis.
Experimental
Materials and reagents
ITO glass was supplied by Zhuhai Kaivo Electronic Components Co. Ltd, China. TiO2 powder (P25) was obtained from Degussa Co. (Germany). Sodium sulfide (Na2S·9H2O) was purchased from Shanghai Asia Chemical Technology Development Co., Ltd (China). Bovine serum albumin (BSA), SA-ALP, and Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) were obtained from Sangon Biotechnology Co., Ltd (Shanghai, China). Normal human serum was provided by Anyan Trade Co., Ltd (Shanghai, China) and used without further treatment. Cadmium nitrate (Cd(NO3)2·4H2O), Tris-(hydroxymethyl)aminomethane (Tris), AA, and manganese acetate (Mn(Ac)2·4H2O) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). AAP and 6-mercaptohexanol (MCH) were provided by Sigma-Aldrich (USA). All other reagents were of analytical grade and used as received. Ultrapure water (18.2 MΩ cm), supplied by the Millipore system (USA), was used throughout. The oligonucleotides used in this work were ordered from Sangon Biotechnology Co., Ltd (Shanghai, China) and the oligonucleotide sequences are listed in Table S1 (see the ESI†).Apparatus
The UV-visible (UV-vis) spectra were obtained via a UV-2100 spectrophotometer (Beijing Lab Tech, China). X-ray diffraction (XRD) analysis was performed on a D8 ADVANCE using Cu Kα radiation (Bruker, Germany). The sample morphology was characterized by scanning electron microscopy (SEM, JSM 6700, Japan) and transmission electron microscopy (TEM, JEM 3010, Japan). A 300 W Xe lamp (Perfect Light, Beijing) with a light intensity of 20 mW cm−2 was used as the irradiation source. All the photoelectrochemical and electrochemical experiments were carried out on an electrochemical workstation (CHI 660D, China) at room temperature with a typical three-electrode system which consisted of a platinum wire (auxiliary electrode), a modified ITO slice (working electrode), and a saturated calomel electrode (SCE, reference electrode).Preparation of the ITO/TiO2/CdS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :Mn electrode
:Mn electrode
Prior to use, the ITO slice (1 × 5 cm) was sonicated in acetone, 1 M NaOH/ethanol (1:1, v/v), and ultrapure water for about 0.25 h, successively, and then dried in an oven at 60 °C for 2 h. After that, 20 μL of 1 mg mL−1 TiO2 was coated on the ITO slice with a fixed area of about 0.24 cm2 (the area of about 0.24 cm2 was fixed through taping a piece of tape with a round hole of 5.5 mm in diameter on the surface of the ITO slice). After being dried in air, the resulting electrode was sintered at 450 °C for 30 min to strengthen the adhesion of TiO2 nanoparticles on the ITO substrate.25,59 Subsequently, due to its good controllability for the preparation process, the successive ionic layer adsorption and reaction (SILAR) method was used to deposit CdS:Mn on the ITO/TiO2 electrode to obtain the ITO/TiO2/CdS:Mn electrode.29,59,60 Briefly, the ITO/TiO2 electrode was firstly dipped into the mixed methanol solution of 0.1 M Cd(NO3)2 and 0.08 M Mn(Ac)2, and then dipped into 0.1 M Na2S methanol solution (1 min each) with intermediate methanol washing until four layers of CdS:Mn were fabricated on the ITO/TiO2 electrode.61
Fabrication of the PEC biosensor and T-DNA assay
Before immobilization on the ITO/TiO2/CdS:Mn electrode, the C-DNA was dissolved in 10 mM Tris-HCl buffer solution (containing 10 mM TCEP, 0.1 M NaCl, pH 7.4) in darkness for 60 min to decrease disulfide bonds. 20 μL C-DNA (1.5 μM) was dripped on the ITO/TiO2/CdS:Mn electrode and incubated at 4 °C for 12 h to immobilize C-DNA on the electrode surface via an S–Cd bond. After rinsing with washing buffer (10 mM Tris-HCl, pH 7.4), the electrode was blocked by 20 μL MCH (2 mM) for 1 h to block the free electrode surface as well as to obtain a well-aligned C-DNA monolayer,62,63 and rinsed thoroughly with washing buffer. Finally, the prepared electrode (ITO/TiO2/CdS:Mn/C-DNA/MCH) was kept at 4 °C for subsequent experiments.
Before CHA reaction, CHA-HP1 and CHA-HP2 (20 mM Tris-HCl solution, 5 mM MgCl2, 100 mM NaCl, pH 7.4) were annealed separately at 90 °C for 5 min and slowly cooled down to room temperature. Then, a mixture of CHA-HP1 (1 μM), CHA-HP2 (1 μM) and T-DNA (different concentrations) was incubated for 2.5 h at 37 °C to conduct the CHA reaction. Subsequently, the mixture solution was transferred to the surface of the prepared electrode and incubation was carried out at 37 °C for 2 h to obtain the ITO/TiO2/CdS:Mn/C-DNA/MCH/CHA-HP1:HP2 electrode. After that, 20 μL of 10 mM Tris-HCl solution (1 mM MgCl2, 500 mM NaCl, pH 7.4) containing 3 μM HCR-HP1-Bio and 3 μM HCR-HP2-Bio was dropped on the electrode surface and incubated at 37 °C for 2 h to perform the HCR process. After rinsing with washing buffer, 20 μL of 1% BSA solution was dropped onto the electrode surface for 30 min to eliminate the possible nonspecific adsorption of protein.64 Subsequently, 20 μL of 0.1 mg mL−1 SA-ALP solution (10 mM Tris-HCl, pH 7.4, 1% BSA) was dripped onto the electrode surface and incubated for 1 h at 37 °C, and then rinsed with washing buffer three times. Finally, the obtained electrode (the ITO/TiO2/CdS:Mn/C-DNA/MCH/CHA-HP1:HP2/HCR-HP1-Bio:HP2-Bio/BSA/SA-ALP electrode, termed FPEC electrode) was incubated in 10 mM Tris-HCl (pH 9.8) containing 0.1 mM Mg(NO3)2 and 10 mM AAP for 1.5 h at 37 °C, and the PEC responses were recorded at the bias potential of −0.3 V.65,66
Results and discussion
Characterization of the photoactive materials
The morphologies of the bare ITO, ITO/TiO2, and ITO/TiO2/CdS:Mn slices were investigated by SEM and TEM. Compared with the bare ITO slice (Fig. 1a), there are a larger number of TiO2 particles on the ITO/TiO2 slice (Fig. 1b) with an average size of about 25 nm based on the TEM image shown in Fig. S1a (see the ESI†). When CdS:Mn is deposited on the ITO/TiO2 slice via the SILAR method, the surface of the ITO/TiO2 slice is changed obviously (Fig. 1c).
 | ||
| Fig. 1 SEM images of (a) bare ITO, (b) ITO/TiO2, and (c) ITO/TiO2/CdS:Mn. | ||
To further verify the successful deposition of CdS:Mn onto the ITO/TiO2 electrode surface, TEM and high-resolution TEM (HR-TEM) images of TiO2/CdS:Mn peeled off from the ITO/TiO2/CdS:Mn slice are shown in Fig. S1b and S1c (see the ESI†). It can be observed that some small nanoparticles exist on the surface of TiO2 nanoparticles (Fig. S1b, see the ESI†). Furthermore, in Fig. S1c (see the ESI†), the lattice spacing of 0.353 nm belongs to the (101) plane of anatase TiO2 (JCPDS no. 21-1272), and the lattice spacing of 0.338 nm can be assigned to the (111) plane of the cubic CdS (JCPDS no. 80-0019). These indicate that CdS is successfully deposited on the surface of TiO2.
Elemental compositions of ITO/TiO2 and ITO/TiO2/CdS:Mn samples were analysed by energy dispersive X-ray spectroscopy (EDS) equipped on a SEM (Fig. 2a). Ti, O, Sn, In, and Si elements exist on both ITO/TiO2 and ITO/TiO2/CdS:Mn samples and should come from the ITO substrate and TiO2 nanoparticles. However, compared with the ITO/TiO2 slice (Fig. 2a, curve a), the ITO/TiO2/CdS:Mn sample has target elements Cd, S, and Mn (curve b), implying that Mn-doped CdS is successfully deposited on the ITO/TiO2 slice. In addition, based on the EDS elemental mappings shown in Fig. S2 (see the ESI†), the elemental distributions of Ti, Cd, and Mn in the ITO/TiO2/CdS:Mn sample are acceptable.
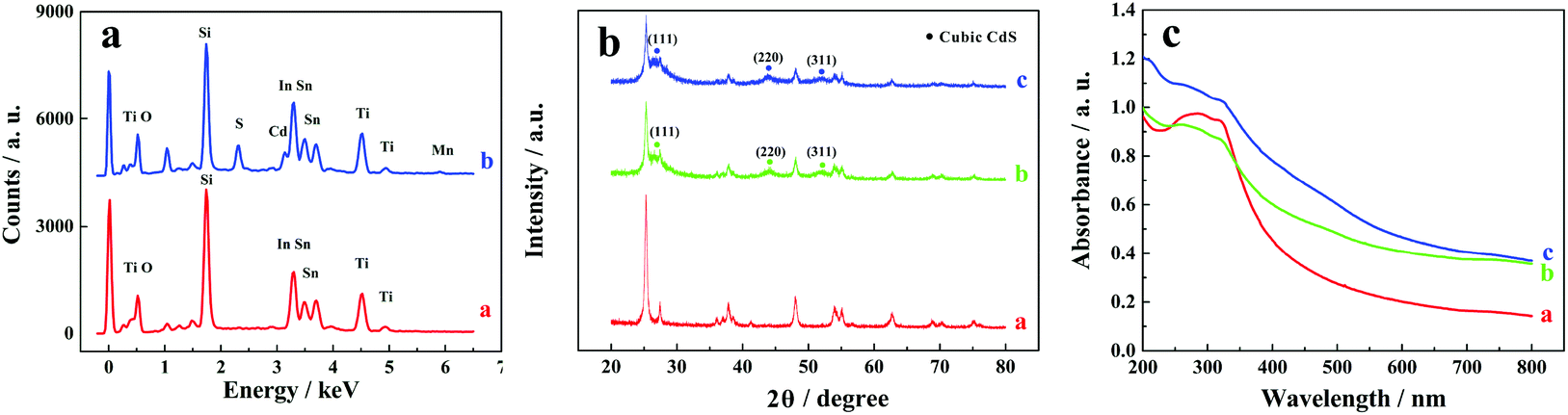 | ||
| Fig. 2 (a) EDS spectra of (curve a) ITO/TiO2 and (curve b) ITO/TiO2/CdS:Mn electrodes, (b) XRD patterns and (c) UV–vis spectra of (curve a) pure TiO2, (curve b) TiO2/CdS, and (curve c) TiO2/CdS:Mn. | ||
The XRD patterns of pure TiO2, TiO2/CdS, and TiO2/CdS:Mn samples are shown in Fig. 2b (the enlarged Fig. 2b is shown in Fig. S3, see the ESI†). From Fig. S3,† all of these patterns show the same peaks of anatase TiO2 and rutile TiO2. Most importantly, three characteristic peaks are observed at 26.51°, 44.08°, and 52.22° in curves b and c (Fig. 2b) and Fig. S3b and S3c (see the ESI†), which correspond to the diffractions of the (111), (220), and (311) planes of the CdS cubic structure (JCPDS no. 80-0019). Furthermore, the XRD pattern of CdS:Mn is almost similar to that of CdS. These results further indicate the successful doping of Mn2+ into CdS nanoparticles and the fabrication of CdS:Mn on the ITO/TiO2 sample.
The UV–vis spectra of pure TiO2, TiO2/CdS and TiO2/CdS:Mn samples are shown in Fig. 2c. It is worth noting that TiO2/CdS (curve b) has a much higher absorption intensity than pure TiO2 (curve a) in the visible light region, revealing that the adsorption properties of TiO2 are enhanced by CdS in the visible light region. In addition, the absorption intensity further increases after the doping of Mn2+ into CdS (curve c). Therefore, CdS:Mn can act as the visible light sensitizer of TiO2 and TiO2/CdS:Mn should be quite suitable as the photoactive material in the PEC assay in this work.
EIS and PEC characterization of the electrode
Electrochemical impedance spectroscopy (EIS) measurements were employed to characterize the interfacial properties and the stepwise modification of the electrode with a redox probe of [Fe(CN)6]3−/4−. In a typical Nyquist plot of the EIS result, the semicircle section at high-frequency corresponds to the charge transfer limited process, and an increase in the related diameter suggests the increase in the interfacial charge transfer resistance (Rct).67 From curve a in Fig. 3a, a small semicircle domain is observed (Rct = 25 Ω), demonstrating that the charge-transfer process of [Fe(CN)6]3−/4− on the bare ITO electrode is fast. After the modification of TiO2/CdS:Mn, the Rct value of the electrode increases, which can be explained as the TiO2/CdS:Mn layer hinders the electron transfer process of [Fe(CN)6]3−/4− at the electrode surface due to the low electric conductivity of the TiO2/CdS:Mn material (curve b). When C-DNA, MCH, CHA-HP1:HP2 (the concentration of T-DNA in the CHA reaction is 20 nM), HCR-HP1-Bio:HP2-Bio, BSA, and SA-ALP are successively introduced onto the electrode surface, the Rct values of the electrode successively increase due to their blocking effect on the electron transfer process of the redox probe at the electrode (from curves c to h).
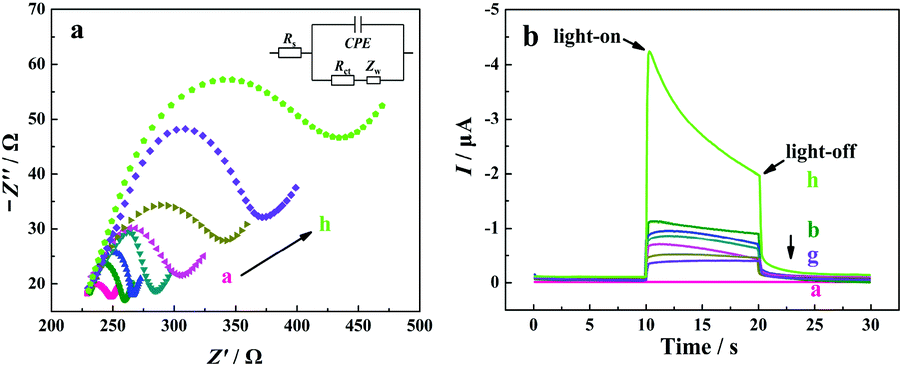 | ||
| Fig. 3 (a) EIS results of the different electrodes in 0.1 M KCl solution with 5 mM [Fe(CN)6]3−/4− (1:1 mixture). Scan rate, 100 mV s−1; amplitude, 5 mV; frequency range, from 0.01 Hz to 100 kHz. Inset plot shows the equivalent electrical circuit. (b) PEC responses of the different electrodes in 10 mM Tris-HCl solution (pH 9.8) containing 20 nM T-DNA, 10 mM AAP and 0.1 mM Mg(NO3)2 at −0.3 V. (a) Bare ITO, (b) ITO/TiO2/CdS:Mn, (c) ITO/TiO2/CdS:Mn/C-DNA, (d) ITO/TiO2/CdS:Mn/C-DNA/MCH, (e) ITO/TiO2/CdS:Mn/C-DNA/MCH/CHA-HP1:HP2, (f) ITO/TiO2/CdS:Mn/C-DNA/MCH/CHA-HP1:HP2/HCR-HP1-Bio:HP2-Bio, (g) ITO/TiO2/CdS:Mn/C-DNA/MCH/CHA-HP1:HP2/HCR-HP1-Bio:HP2-Bio/BSA, (h) ITO/TiO2/CdS:Mn/C-DNA/MCH/CHA-HP1:HP2/HCR-HP1-Bio:HP2-Bio/BSA/SA-ALP. | ||
The fabrication process of the “signal-on” PEC biosensor was also monitored by photocurrent response in 10 mM Tris-HCl solution (pH 9.8) containing 0.1 mM Mg(NO3)2 and 10 mM AAP at −0.3 V with the catalysis reaction time of 1.5 h. As shown in Fig. 3b, for the bare ITO electrode, no photocurrent response is observed (curve a). After the coating of TiO2/CdS:Mn nanocomposite onto the ITO surface, a strong photocurrent is observed (curve b, −0.98 μA), indicating that CdS:Mn/TiO2 is a promising photoelectric conversion material. After that, with the successive introduction of C-DNA, MCH, CHA-HP1:HP2, HCR-HP1-Bio:HP2-Bio, and BSA onto the surface of the ITO/TiO2/CdS:Mn electrode, the photocurrents successively decrease (from curves c to g), which corresponds to the EIS results. However, when SA-ALP is further introduced onto the electrode surface and incubated for 1.5 h at 37 °C for in situ catalytic generation of AA from AAP, the photocurrent response of the electrode largely increases (curve h). The obvious increase of the photocurrent should be due to the excellent electron donor (AA), which is confirmed by the results shown in Fig. S4 (see the ESI†). From Fig. S4,† the photocurrent of the FPEC electrode in 1 mM AA solution is about ten times larger than that in the same solution without AA. On the other hand, it is noted that the photocurrent decays with the increase in the illumination time due to the consumption of AA. In light of all these results, the developed “signal-on” PEC biosensor should be feasible for T-DNA assay. All of the results demonstrate the successful fabrication of the developed PEC biosensor based on the three-stage cascade signal amplification strategy according to the procedure shown in Scheme 1b.
Optimization of the experimental conditions
The incubation time for CHA, applied potential, the concentrations of C-DNA, SA-ALP and AAP, and the catalysis reaction time between ALP and AAP are the important factors which may affect the analysis capability and optimum state of the proposed “signal-on” PEC biosensor. Therefore, a series of control experiments were carried out.The incubation time for CHA influences the amount of the CHA-HP1:HP2 duplex immobilized on the electrode surface, and indirectly influences the amount of the ALP enzyme. Therefore, the optimal incubation time for CHA was investigated by recording the photocurrent intensity of the FPEC electrode with a series of different incubation times. As shown in Fig. S5 (see the ESI†), the photocurrent intensity increases and reaches a platform at 2.5 h, revealing that the reaction equilibrium for the CHA process is achieved. Therefore, 2.5 h is the appropriate incubation time for CHA in the following experiments.
As another important factor, the effect of the applied potential on the photocurrent intensity of the FPEC electrode was investigated and the results are shown in Fig. S6 (see the ESI†). The anodic photocurrents are very small from −0.1 to 0.3 V. However, the cathodic photocurrents are much larger than the anodic photocurrents and increase obviously from −0.2 to −0.4 V. It is noted that the photocurrent at −0.3 V is about 76.8% of that at −0.4 V and the sensitivity of the developed PEC sensor at −0.3 V is acceptable. Considering the bad effects of the high applied potential (either anodic or cathodic potential) on the biomolecules and the surface state of the photoelectrodes,68 −0.3 V is selected as the optimal applied potential for T-DNA assay.
The effect of the C-DNA concentration on the photocurrent intensity of the FPEC electrode was also evaluated. With the increase of the C-DNA concentration, the photocurrent intensity increases and a maximum value is obtained at 1.5 μM C-DNA (Fig. S7, see the ESI†). When the C-DNA concentration is more than 1.5 μM, the photocurrent intensity of the FPEC electrode decreases due to the possible reason that the excessive amount of C-DNA decreases the hybridization efficiency between C-DNA and the CHA-HP1/CHA-HP2 complex. Therefore, 1.5 μM is selected as the optimum concentration of C-DNA.
Based on Scheme 1a and the results shown in Fig. 3b, the amount of the in situ generated AA is the key factor affecting the assay performance of the developed PEC biosensor. Therefore, the related parameters (the concentrations of ALP enzyme and its substrate AAP, the reaction time between ALP and AAP) should be optimized. From Fig. S8 to S10 (see the ESI†), the optimal values of these parameters (ALP concentration, 0.1 mg mL−1; AAP concentration, 10 mM; the reaction time between ALP and AAP, 1.5 h) are obtained. When the values of these parameters are larger than their optimal values, plateaus are reached due to the related saturated states.
PEC assay of T-DNA
Under the optimized experimental conditions, photocurrent responses of the “signal-on” PEC biosensor to different T-DNA concentrations have been investigated in 10 mM Tris-HCl solution (pH 9.8) containing 10 mM AAP and 0.1 mM Mg(NO3)2 and the related results are shown in Fig. 4a. It is noted that the photocurrent increases with the increase of the T-DNA concentration in the range from 0 to 20 nM. Fig. 4b shows the relationship between the photocurrent and the logarithm of the T-DNA concentration. The photocurrent is linear with the T-DNA concentration ranging from 0.1 fM to 100 pM with the linear regression equation of I (μA) = −0.3536logCT (fM) − 2.2472 (R2 = 0.9963). According to the signal-to-noise ratio of 3 and the photocurrent in blank solution (−1.775 μA), the detection limit is calculated to be 0.052 fM. It is worth noting that the developed “signal-on” PEC biosensor based on the three-stage cascade signal amplification strategy exhibits a wider linear range and a lower detection limit than most of the reported methods (Table S2, see the ESI†).
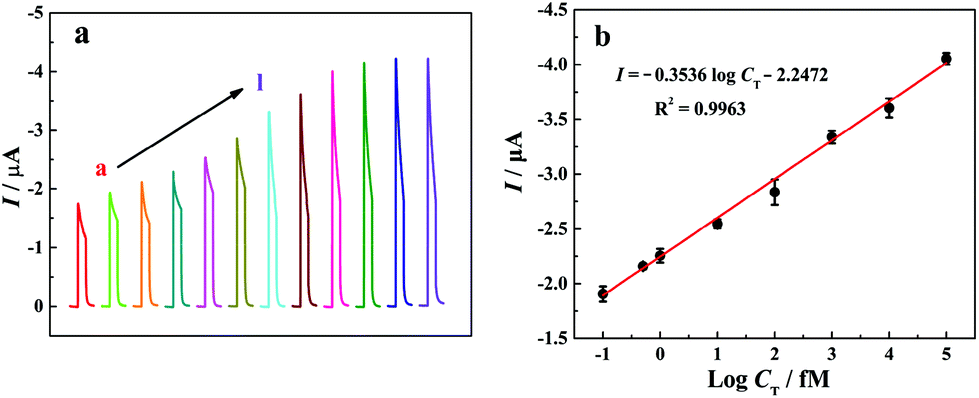 | ||
| Fig. 4 (a) Photocurrent responses of the “signal-on” PEC biosensor in 10 mM Tris-HCl solution (pH 9.8) containing 10 mM AAP and different concentrations of T-DNA (from a to l): 0 fM, 0.1 fM, 0.5 fM, 1 fM, 10 fM, 100 fM, 1 pM, 10 pM, 100 pM, 1 nM, 10 nM, and 20 nM. (b) The linear calibration curve of the “signal-on” PEC biosensor for T-DNA detection. Error bars represent the standard deviation of three parallel experiments. | ||
Stability, reproducibility, and selectivity of the PEC biosensor
Stability is an important parameter for the PEC biosensor. After the ITO/TiO2/CdS:Mn/C-DNA/MCH electrode is stored in a refrigerator at 4 °C for two weeks, the PEC response still remains at 96.1% of the initial intensity for 1 pM T-DNA detection. The result reveals that the stability of the developed PEC system is acceptable. Moreover, the reproducibility of the proposed “signal-on” PEC biosensor was evaluated, and the relative standard deviation (RSD) for T-DNA (1 pM) assay on five modified electrodes is 5.12%. This indicates that the proposed “signal-on” PEC biosensor has a good reproducibility for T-DNA detection.
The selectivity of the developed “signal-on” PEC biosensor was also evaluated. Four kinds of DNA sequences (T-DNA, Sm-DNA (single-base mismatched DNA), Tm-DNA (three-base mismatched DNA), and N-DNA (noncomplementary DNA)) were designed and detected with the same concentration (1 pM). As shown in Fig. 5, the photocurrent responses |ΔI| (ΔI = I − I0, I0 and I stand for the photocurrents of the developed PEC biosensor in the absence and presence of different DNA sequences) for N-DNA, Tm-DNA, Sm-DNA and T-DNA are 0.093 μA, 0.463 μA, 0.667 μA and 1.537 μA, respectively. It is noted that the photocurrent response for Tm-DNA (Sm-DNA) is only about 30.1% (43.4%) of that for T-DNA. These results are basically consistent with those reported in previous papers,12,69 indicating that the developed “signal-on” PEC biosensor exhibits an acceptable selectivity to distinguish the perfect complementary DNA and the base mismatched DNA.
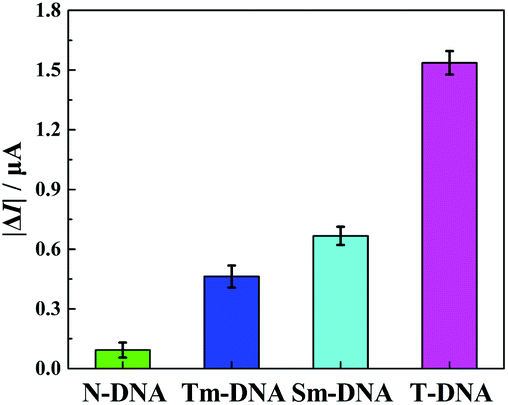 | ||
| Fig. 5 Selectivity of the developed PEC biosensor toward 1 pM of N-DNA, Tm-DNA, Sm-DNA, and T-DNA in 10 mM Tris-HCl solution (10 mM AAP, 0.1 mM Mg(NO3)2, pH 9.8). Error bars represent the standard deviation of three parallel experiments. | ||
Recovery test
In order to verify the applicability and reliability of the developed PEC biosensor, different concentrations of T-DNA (100 fM, 500 fM, 1000 fM, and 5000 fM) were added into the 10-fold diluted human blood serum samples which were diluted by 10 mM Tris-HCl buffer (pH 7.4), and the photocurrent measurement was performed in 10 mM Tris-HCl solution (10 mM AAP, 0.1 mM Mg(NO3)2, pH 9.8). The average recoveries for the added T-DNA with 100 fM, 500 fM, 1000 fM, and 5000 fM are 94.9%, 106.2%, 96.5%, and 104.0%, respectively (Table 1), indicating that the recovery of the developed “signal-on” PEC biosensor is acceptable and the proposed method may have potential application in T-DNA assay in real samples.| Sample | Added (fM) | Found (fM) | RSD | Recovery (%) |
|---|---|---|---|---|
| a Abbreviation: relative standard deviation (RSD). | ||||
| 1 | 100 | 94.9 ± 6.0 | 4.62% | 94.9 |
| 2 | 500 | 530.9 ± 31.5 | 4.32% | 106.2 |
| 3 | 1000 | 964.7 ± 70.1 | 5.31% | 96.5 |
| 4 | 5000 | 5214.8 ± 126 | 4.92% | 104.0 |
Conclusions
A novel and ultrasensitive “signal-on” PEC biosensor has been developed for T-DNA assay based on a three-stage cascade signal amplification strategy by combining CHA, HCR and enzyme catalytic amplification methods. Due to the signal amplification elements, the methods of CHA and HCR provide plenty of biotins to anchor sufficient SA-ALP molecules which catalyze AAP to in situ generate AA (electron donor), resulting in an enhanced photocurrent response. The developed “signal-on” PEC biosensor shows superior analytical performance for sensitive determination of T-DNA with a detection limit as low as 0.052 fM, and could be an attractive candidate in bioanalysis, clinical biomedicine, and disease diagnostics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the NSFC (21475035, 21727810), the Foundation for Innovative Research Groups of NSFC (21521063) and the China Scholarship Council (201606130011).Notes and references
- A. Baeissa, N. Dave, B. D. Smith and J. W. Liu, ACS Appl. Mater. Interfaces, 2010, 2, 3594–3600 Search PubMed.
- Y. Y. Zhang, Z. W. Tang, J. Wang, H. Wu, A. H. Maham and Y. Lin, Anal. Chem., 2010, 82, 6440–6446 CrossRef PubMed.
- E. H. Xiong, X. X. Yan, X. H. Zhang, Y. Q. Liu, J. W. Zhou and J. H. Chen, Biosens. Bioelectron., 2017, 87, 732–736 CrossRef PubMed.
- E. H. Xiong, X. H. Zhang, Y. Q. Liu, J. W. Zhou, P. Yu, X. Y. Li and J. H. Chen, Anal. Chem., 2015, 87, 7291–7296 CrossRef PubMed.
- E. H. Xiong, Z. Z. Li, X. H. Zhang, J. W. Zhou, X. X. Yan, Y. Q. Liu and J. H. Chen, Anal. Chem., 2017, 89, 8830–8835 CrossRef PubMed.
- J. H. Huang, X. F. Su and Z. G. Li, Anal. Chem., 2012, 84, 5939–5943 CrossRef PubMed.
- G. L. Liu, J. J. Li, D. Q. Feng, J. J. Zhu and W. Wang, Anal. Chem., 2016, 89, 1002–1008 CrossRef PubMed.
- S. F. Liu, C. X. Zhang, J. J. Ming, C. F. Wang, T. Liu and F. Li, Chem. Commun., 2013, 49, 7947–7949 RSC.
- R. Hu, T. Liu, X. B. Zhang, S. Y. Huan, C. C. Wu, T. Fu and W. H. Tan, Anal. Chem., 2014, 86, 5009–5016 CrossRef PubMed.
- J. A. He, F. Zhao, C. L. Wu, J. Y. Yao, L. Shi, C. X. Liu, C. Z. Zhao, Y. Q. Xu, X. A. Wang and D. Y. Gu, J. Mater. Chem. B, 2013, 1, 5398–5402 RSC.
- W. Diao, M. Tang, S. J. Ding, X. M. Li, W. B. Cheng, F. Mo, X. Y. Yan, H. M. Ma and Y. R. Yan, Biosens. Bioelectron., 2018, 100, 228–234 CrossRef PubMed.
- Y. Gao and B. X. Li, Anal. Chem., 2013, 85, 11494–11500 CrossRef PubMed.
- Y. Gao and B. X. Li, Anal. Chem., 2014, 86, 8881–8887 CrossRef PubMed.
- H. R. Zhang, J. J. Xu and H. Y. Chen, Anal. Chem., 2013, 85, 5321–5325 CrossRef PubMed.
- Y. Chai, D. Y. Tian, W. Wang and H. Cui, Chem. Commun., 2010, 46, 7560–7562 RSC.
- Y. Zang, J. P. Lei, L. Zhang and H. X. Ju, Anal. Chem., 2014, 86, 12362–12368 CrossRef PubMed.
- S. L. Liu, C. Li, J. Cheng and Y. X. Zhou, Anal. Chem., 2006, 78, 4722–4726 CrossRef PubMed.
- C. X. Li, H. Y. Wang, J. Shen and B. Tang, Anal. Chem., 2015, 87, 4283–4291 CrossRef PubMed.
- T. Hou, L. F. Zhang, X. Z. Sun and F. Li, Biosens. Bioelectron., 2016, 75, 359–364 CrossRef PubMed.
- J. X. Chen and G. C. Zhao, Biosens. Bioelectron., 2017, 98, 155–160 CrossRef PubMed.
- N. Haddour, J. Chauvin, C. Gondran and S. Cosnier, J. Am. Chem. Soc., 2006, 128, 9693–9698 CrossRef PubMed.
- Y. F. Peng, G. S. Yi and Z. Q. Gao, Chem. Commun., 2010, 46, 9131–9133 RSC.
- H. Zhu, G. C. Fan, E. S. Abdel-Halim, J. R. Zhang and J. J. Zhu, Biosens. Bioelectron., 2016, 77, 339–346 CrossRef PubMed.
- Y. J. Li, M. J. Ma and J. J. Zhu, Anal. Chem., 2012, 84, 10492–10499 CrossRef PubMed.
- G. C. Fan, M. Zhao, H. Zhu, J. J. Shi, J. R. Zhang and J. J. Zhu, J. Phys. Chem. C, 2015, 120, 15657–15665 Search PubMed.
- L. M. Pan, Q. J. He, J. N. Liu, Y. Chen, M. Ma, L. L. Zhang and J. L. Shi, J. Am. Chem. Soc., 2012, 134, 5722–5725 CrossRef PubMed.
- Y. L. Lee, C. F. Chi and S. Y. Liau, Chem. Mater., 2009, 22, 922–927 CrossRef.
- N. Pradhan and D. D. Sarma, J. Phys. Chem. Lett., 2011, 2, 2818–2826 CrossRef.
- G. C. Fan, H. Zhu, D. Du, J. R. Zhang, J. J. Zhu and Y. H. Lin, Anal. Chem., 2016, 88, 3392–3399 CrossRef PubMed.
- G. C. Fan, L. Han, H. Zhu, J. R. Zhang and J. J. Zhu, Anal. Chem., 2014, 86, 12398–12405 CrossRef PubMed.
- S. W. Zhou, Y. Y. Wang, M. Zhao, L. P. Jiang and J. J. Zhu, ChemPhysChem, 2015, 16, 2826–2835 CrossRef.
- Z. L. Ge, M. H. Lin, P. Wang, H. Pei, J. Yan, J. Y. Shi, Q. Huang, D. N. He, C. H. Fan and X. L. Zuo, Anal. Chem., 2014, 86, 2124–2130 CrossRef PubMed.
- Y. Chen, J. Xu, J. Su, Y. Xiang, R. Yuan and Y. Q. Chai, Anal. Chem., 2012, 84, 7750–7755 CrossRef PubMed.
- G. H. Yao, R. P. Liang, X. D. Yu, C. F. Huang, L. Zhang and J. D. Qiu, Anal. Chem., 2014, 87, 929–936 CrossRef PubMed.
- W. Shen, H. M. Deng and Z. Q. Gao, J. Am. Chem. Soc., 2012, 134, 14678–14681 CrossRef PubMed.
- H. Deng, Y. Xu, Y. H. Liu, Z. J. Che, H. L. Guo, S. X. Shan, Y. Sun, X. F. Liu, K. Y. Huang and X. W. Ma, Anal. Chem., 2012, 84, 1253–1258 CrossRef PubMed.
- K. Y. Zhang, S. Z. Lv, Z. Z. Lin, M. J. Li and D. P. Tang, Biosens. Bioelectron., 2018, 101, 159–166 CrossRef PubMed.
- H. Yan, Y. C. Xu, Y. Lu and W. L. Xing, Anal. Chem., 2017, 89, 10137–10140 CrossRef PubMed.
- L. Ge, X. M. Sun, Q. Hong and F. Li, ACS Appl. Mater. Interfaces, 2017, 9, 32089–32096 Search PubMed.
- X. Hun, Y. Meng, S. S. Wang, H. Zhang and X. L. Luo, Sens. Actuators, B, 2018, 254, 347–353 CrossRef.
- Y. Wang, N. Gan, Y. Zhou, T. H. Li, F. T. Hu, Y. T. Cao and Y. J. Chen, Biosens. Bioelectron., 2017, 97, 100–106 CrossRef PubMed.
- F. Kivlehan, F. Mavré, L. Talini, B. Limoges and D. Marchal, Analyst, 2011, 136, 3635–3642 RSC.
- F. Ma, M. Liu, B. Tang and Y. Zhang, Anal. Chem., 2017, 89, 6182–6187 CrossRef PubMed.
- M. Jiao, G. F. Jie, L. Tan and S. Y. Niu, Anal. Chim. Acta, 2017, 983, 166–172 CrossRef PubMed.
- D. Yin, Y. Y. Tao, L. Tang, W. Li, Z. Zhang, J. L. Li and G. M. Xie, Microchim. Acta, 2017, 184, 3721–3728 CrossRef.
- P. Yin, H. M. Choi, C. R. Calvert and N. A. Pierce, Nature, 2008, 451, 318–322 CrossRef PubMed.
- X. Chen, J. Am. Chem. Soc., 2011, 134, 263–271 CrossRef PubMed.
- Y. S. Jiang, S. Bhadra, B. L. Li and A. D. Ellington, Angew. Chem., Int. Ed., 2014, 126, 1876–1879 CrossRef.
- B. L. Li, Y. Jiang, X. Chen and A. D. Ellington, J. Am. Chem. Soc., 2012, 134, 13918–13921 CrossRef PubMed.
- L. Q. Zhang, Y. J. Liu, Y. Li, Y. W. Zhao, W. Wei and S. Q. Liu, Anal. Chim. Acta, 2016, 933, 75–81 CrossRef PubMed.
- Y. J. Jeong, K. Park and D. E. Kim, Cell. Mol. Life Sci., 2009, 66, 3325–3336 CrossRef PubMed.
- Y. Zang, J. P. Lei, P. H. Ling and H. X. Ju, Anal. Chem., 2015, 87, 5430–5436 CrossRef PubMed.
- L. Ge, W. X. Wang, T. Hou and F. Li, Biosens. Bioelectron., 2016, 77, 220–226 CrossRef PubMed.
- J. Yoshizumi, S. Kumamoto, M. Nakamura and K. Yamana, Analyst, 2008, 133, 323–325 RSC.
- D. F. Roucoux and E. L. Murphy, AIDS Rev., 2004, 6, 144–154 Search PubMed.
- Q. M. Shen, L. Han, G. C. Fan, J. R. Zhang, L. P. Jiang and J. J. Zhu, Anal. Chem., 2015, 87, 4949–4956 CrossRef PubMed.
- S. W. Zhou, Y. Kong, Q. M. Shen, X. L. Ren, J. R. Zhang and J. J. Zhu, Anal. Chem., 2014, 86, 11680–11689 CrossRef PubMed.
- W. W. Zhao, Z. Y. Ma, D. Y. Yan, J. J. Xu and H. Y. Chen, Anal. Chem., 2012, 84, 10518–10521 CrossRef PubMed.
- G. C. Fan, L. Han, J. R. Zhang and J. J. Zhu, Anal. Chem., 2014, 86, 10877–10884 CrossRef PubMed.
- A. R. Liu, K. F. Yin, L. Mi, M. Y. Ma, Y. J. Liu, Y. Li, W. Wei, Y. J. Zhang and S. Q. Liu, Anal. Chim. Acta, 2017, 973, 82–90 CrossRef PubMed.
- J. Song, J. M. Wang, X. Y. Wang, W. Zhao, Y. Q. Zhao, S. Wu, Z. M. Gao, J. L. Yuan and C. G. Meng, Biosens. Bioelectron., 2016, 80, 614–620 CrossRef PubMed.
- C. Li, Y. Q. Tao, Y. Yang, Y. Xiang and G. X. Li, Anal. Chem., 2017, 89, 5003–5007 CrossRef PubMed.
- F. L. Gao, T. T. Fan, S. S. Ou, J. Wu, X. Zhang, J. J. Luo, N. Li, Y. Yao, Y. F. Mou, X. J. Liao and D. Q. Geng, Biosens. Bioelectron., 2018, 99, 201–208 CrossRef PubMed.
- C. Wang, H. Zhou, W. P. Zhu, H. B. Li, J. H. Jiang, G. L. Shen and R. Q. Yu, Biosens. Bioelectron., 2013, 47, 324–328 CrossRef PubMed.
- X. Li, L. S. Zhu, Y. L. Zhou, H. S. Yin and S. Y. Ai, Anal. Chem., 2017, 89, 2369–2376 CrossRef PubMed.
- H. S. Yin, B. Sun, L. F. Dong, B. C. Li, Y. L. Zhou and S. Y. Ai, Biosens. Bioelectron., 2015, 64, 462–468 CrossRef PubMed.
- Y. Cao, S. Zhu, J. C. Yu, X. J. Zhu, Y. M. Yin and G. X. Li, Anal. Chem., 2012, 84, 4314–4320 CrossRef PubMed.
- N. Zhang, Z. Y. Ma, Y. F. Ruan, W. W. Zhao, J. J. Xu and H. Y. Chen, Anal. Chem., 2016, 88, 1990–1994 CrossRef PubMed.
- F. Xuan, X. T. Luo and I. M. Hsing, Anal. Chem., 2012, 84, 5216–5220 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Sequences of oligonucleotides; TEM images of pure TiO2 and TiO2/CdS:Mn; EDS mapping of the ITO/TiO2/CdS:Mn sample; XRD patterns of TiO2, TiO2/CdS, and TiO2/CdS:Mn; the role of AA in the PEC response of the FPEC electrode; optimization of the experimental conditions; comparison of various analytical methods for DNA detection. See DOI: 10.1039/c8an00609a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |