DOI:
10.1039/C7AN01984J
(Paper)
Analyst, 2018,
143, 1250-1258
Simultaneous non-polar and polar lipid analysis by on-line combination of HILIC, RP and high resolution MS†
Received
8th December 2017
, Accepted 5th February 2018
First published on 5th February 2018
Abstract
Given the chemical diversity of lipids and their biological relevance, suitable methods for lipid profiling and quantification are demanded to reduce sample complexity and analysis times. In this work, we present a novel on-line chromatographic method coupling hydrophilic interaction liquid chromatography (HILIC) dedicated to class-specific separation of polar lipid to reversed-phase chromatography (RP) for non-polar lipid analysis. More specifically, the void volume of the HILIC separation-consisting of non-polar lipids- is transferred to the orthogonal RP column enabling the on-line combination of HILIC with RP without any dilution in the second dimension. In this setup the orthogonal HILIC and RP separations were performed in parallel and the effluents of both columns were combined prior to high-resolution MS detection, offering the full separation space in one analytical run. Rapid separation for both polar and non-polar lipids within only 15 min (including reequilibration time) was enabled using sub-2 μm particles and UHPLC. The method proved to be robust with excellent retention time stability (RSDs < 1%) and LODs in the fmol to pmol (absolute on column) range even in the presence of complex biological matrix such as human plasma. The presented high-resolution LC-MS/MS method leads to class-specific separation of polar lipids and separation of non-polar lipids which is lost in conventional HILIC separations. HILIC-RP-MS is a promising tool for targeted and untargeted lipidomics workflows as three interesting features are combined namely (1) the decreased run time of state of the art shotgun MS methods, (2) the elevated linear dynamic range inherent to chromatographic separation and (3) increased level of identification by separation of polar and non-polar lipid classes.
1. Introduction
Lipids can be classified into categories by their chemical and structural similarity,1 they can be grouped into polar and non-polar lipids based on their overall hydrophobicity or categorized by their molecular building blocks.2 Given the extremely high diversity of lipids (over 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 unique lipid structures annotated in the Lipid Maps Structure Database3,4) and increasing proof of their biological relevance,3,5–9 the urge to develop novel methods for lipid profiling and quantification continues with the major aim to reduce sample complexity and analysis times. High-resolution mass spectrometry (HRMS) has evolved as a key technique in lipidomics as it provides lipid identification by accurate mass and fragmentation pattern at the same time allowing to cope with complex samples.10 On general terms, MS based lipidomics strategies involve (1) direct-infusion shotgun lipidomics approaches10–12 and/or (2) the combination of liquid chromatography (LC) and MS.2,13–18 Shotgun lipidomics offers the advantage of fast lipid profiling but the direct infusion leads to problems with isomeric and isobaric lipid species and a limited dynamic range in a sample of interest. LC-MS based approaches offer (1) an increased dynamic range and (2) an additional level of identification by retention time. Different chromatographic separations were developed for lipidomics tasks including reversed-phase chromatography (RP), normal phase chromatography (NP), hydrophilic interaction chromatography (HILIC), strong anion exchange chromatography (SAX) and supercritical fluid chromatography (SFC).19–25 Indeed, RP chromatography separates lipids based on hydrophobic properties such as fatty acid chain length, degree of saturation and double bond position. RP chromatography has emerged as state of the art method for LC-MS based lipidomics analysis as it allows polar and non-polar lipid analysis in one run, however isobaric overlaps from different lipid classes can occur complicating lipid identification.13,19,22,26–29 HILIC chromatography on the other hand avoids isobaric overlaps between different classes as it enables lipid class-separation by hydrophilic interaction. Lipid classes share a common hydrophilic headgroup so that hydrophobic differences of lipids within one class can be neglected in HILIC chromatography. Hence, class-specific coelution of all lipids occurs in HILIC and was successfully applied for polar lipid analysis e.g. glycerophospholipids, glycosphingolipids and phosphosphingolipids.16,23,28,30–32 HILIC and RP chromatography as well as their combinations proved to be extremely powerful for lipid analysis in many previous studies.13,14,22,27,28,30–36 With the advent of the UHPLC technology and the downscaling of chromatographic filling material by sub-2 μm particles or fused-core particles, increased plate numbers and enhanced chromatographic speed are enabled.37–41 However, implementation of orthogonal RP and HILIC into 2D approaches is challenging due to incompatible eluent systems. Solutions for this problem are (1) dilution of the incompatible mobile phase, (2) use of a trapping column (3) minimizing the transferred volume between both dimensions or (4) offline combinations of HILIC and RP.21,28,32,36,42–45 In this work a novel UHPLC based workflow for the direct combination of a short HILIC column (sub-2 μm) in the first dimension with a RP column (sub-2 μm) in the second dimension is tested for the simultaneous analysis of polar and non-polar lipids. Classical 2D multiple heart-cutting approaches transfer several chromatographic peaks from one dimension to the next in order to perform comprehensive analysis on coeluting species exploiting complementary stationary phase interactions.21,32,42,46–51 Here, we aim to transfer only the void volume (one fraction) of the HILIC column to the RP column to obtain class-specific retentivity of polar lipids at the same time retaining non-polar lipids in one analytical run. Moreover, the HILIC-RP-HRMS workflow is tested for untargeted and targeted lipidomics applications.
000 unique lipid structures annotated in the Lipid Maps Structure Database3,4) and increasing proof of their biological relevance,3,5–9 the urge to develop novel methods for lipid profiling and quantification continues with the major aim to reduce sample complexity and analysis times. High-resolution mass spectrometry (HRMS) has evolved as a key technique in lipidomics as it provides lipid identification by accurate mass and fragmentation pattern at the same time allowing to cope with complex samples.10 On general terms, MS based lipidomics strategies involve (1) direct-infusion shotgun lipidomics approaches10–12 and/or (2) the combination of liquid chromatography (LC) and MS.2,13–18 Shotgun lipidomics offers the advantage of fast lipid profiling but the direct infusion leads to problems with isomeric and isobaric lipid species and a limited dynamic range in a sample of interest. LC-MS based approaches offer (1) an increased dynamic range and (2) an additional level of identification by retention time. Different chromatographic separations were developed for lipidomics tasks including reversed-phase chromatography (RP), normal phase chromatography (NP), hydrophilic interaction chromatography (HILIC), strong anion exchange chromatography (SAX) and supercritical fluid chromatography (SFC).19–25 Indeed, RP chromatography separates lipids based on hydrophobic properties such as fatty acid chain length, degree of saturation and double bond position. RP chromatography has emerged as state of the art method for LC-MS based lipidomics analysis as it allows polar and non-polar lipid analysis in one run, however isobaric overlaps from different lipid classes can occur complicating lipid identification.13,19,22,26–29 HILIC chromatography on the other hand avoids isobaric overlaps between different classes as it enables lipid class-separation by hydrophilic interaction. Lipid classes share a common hydrophilic headgroup so that hydrophobic differences of lipids within one class can be neglected in HILIC chromatography. Hence, class-specific coelution of all lipids occurs in HILIC and was successfully applied for polar lipid analysis e.g. glycerophospholipids, glycosphingolipids and phosphosphingolipids.16,23,28,30–32 HILIC and RP chromatography as well as their combinations proved to be extremely powerful for lipid analysis in many previous studies.13,14,22,27,28,30–36 With the advent of the UHPLC technology and the downscaling of chromatographic filling material by sub-2 μm particles or fused-core particles, increased plate numbers and enhanced chromatographic speed are enabled.37–41 However, implementation of orthogonal RP and HILIC into 2D approaches is challenging due to incompatible eluent systems. Solutions for this problem are (1) dilution of the incompatible mobile phase, (2) use of a trapping column (3) minimizing the transferred volume between both dimensions or (4) offline combinations of HILIC and RP.21,28,32,36,42–45 In this work a novel UHPLC based workflow for the direct combination of a short HILIC column (sub-2 μm) in the first dimension with a RP column (sub-2 μm) in the second dimension is tested for the simultaneous analysis of polar and non-polar lipids. Classical 2D multiple heart-cutting approaches transfer several chromatographic peaks from one dimension to the next in order to perform comprehensive analysis on coeluting species exploiting complementary stationary phase interactions.21,32,42,46–51 Here, we aim to transfer only the void volume (one fraction) of the HILIC column to the RP column to obtain class-specific retentivity of polar lipids at the same time retaining non-polar lipids in one analytical run. Moreover, the HILIC-RP-HRMS workflow is tested for untargeted and targeted lipidomics applications.
2. Materials and methods
2.1 Lipid standards
The majority of lipid standards (LPC 18:0, LPC 16:0, DG 34:1, TG 52:2, PA 36:2, PC 34:1, PC 34:2, PC O-36:2, PE 36:2, PG 36:2, PI 34:1, PS 36:2, HexCer d34:1, Cer d36:1, SM d42:2, Hex2Cer d34:1) were obtained from Avanti Polar Lipids, Inc. (Alabaster, Alabama, USA) and some lipids (FA 16:0, FA 18:0, CE 18:0, CE 18:2, ST 28:3/ergosterol) were ordered at Sigma Aldrich (Vienna, Austria).
2.2 Lipid extraction
The yeast samples were fermented and extracted as previously described34,52 with chloroform/methanol (2:1, v/v) applying the Folch protocol.53 Yeast extract aliquots (n = 3) of 500 μL corresponding to 107 cells were processed. The certified reference material SRM 1950 was ordered at NIST (Gaithersburg, USA) and 100 μL aliquots (n = 3) were extracted by MTBE using the Matyash protocol.54 All samples were dried under reduced pressure after extraction and reconstituted in 200 μL 95% ACN and 5% 40 mM ammonium formate (in H2O, pH = 4.0) for yeast and 400 μL for plasma samples.
2.3 Hydrophilic interaction liquid chromatography (HILIC)
An Acquity UPLC BEH Amide (2.1 × 100 mm, 130 Å pore size, Waters) was used employing sub 2 μm silica particles (1.7 μm particle size) for hydrophilic interaction liquid chromatography. The column was used in combination with a preheater and a Viper UHPLC inline filter (Thermo Fisher Scientific). The flow rate was set to 250 μL min−1 and the column temperature to 40 °C. All mobile phase solvents were of LC-MS grade and ordered at Fisher Scientific (Vienna, Austria) or Sigma Aldrich (Vienna, Austria). The mobile phase A consisted of 40 mM ammonium formate (pH = 4.0) and mobile phase B was 100% acetonitrile (ACN) using the following gradient: 0–2.0 min 96% B, 2.0–7.0 min ramp to 70% B, 7.0–7.5 min 70% B, 7.5–15.0 min-fast switch to 96% B and 7.5 min long equilibration step. The injection volume was 2 μL (MS1 quantification runs) and 10 μL (ddMS2 identification runs) and the injector needle was washed with 75% isopropanol (IPA), 25% H2O, 0.1% formic acid prior to each injection to remove excess lipids from the injection system.
2.4 Reversed-phase (RP) chromatography
A C18 Acquity UHPLC HSS T3 (2.1 mm × 150 mm, 100 Å pore size, 1.8 μm, Waters) was used for second-dimension separation. The column was fitted with a VanGuard Pre-column, (2.1 mm × 5 mm, 100 Å, 1.8 μm). The flow rate was set to 250 μL min−1 and the column temperature to 40 °C. All mobile phase solvent were of LC-MS grade and ordered at Fisher Scientific or Sigma Aldrich. Solvent A was ACN/H2O (3:2, v/v), and solvent B was IPA/ACN (9:1, v/v). Both solvents contained 0.1% formic acid and 10 mM ammonium formate. The following gradient was used: 0–4.5 min 70% B, 4.5–6.0 min ramp to 100% B, 6.0–9.0 min 100% B, 9.0–15.0 min-fast switch to 30% B for a 6 min long equilibration step. The injection volume was 2 μL and the injector needle was washed with 75% IPA, 25% H2O, 0.1% formic acid prior to each injection.
2.5 Setup for HILIC-RP liquid chromatography
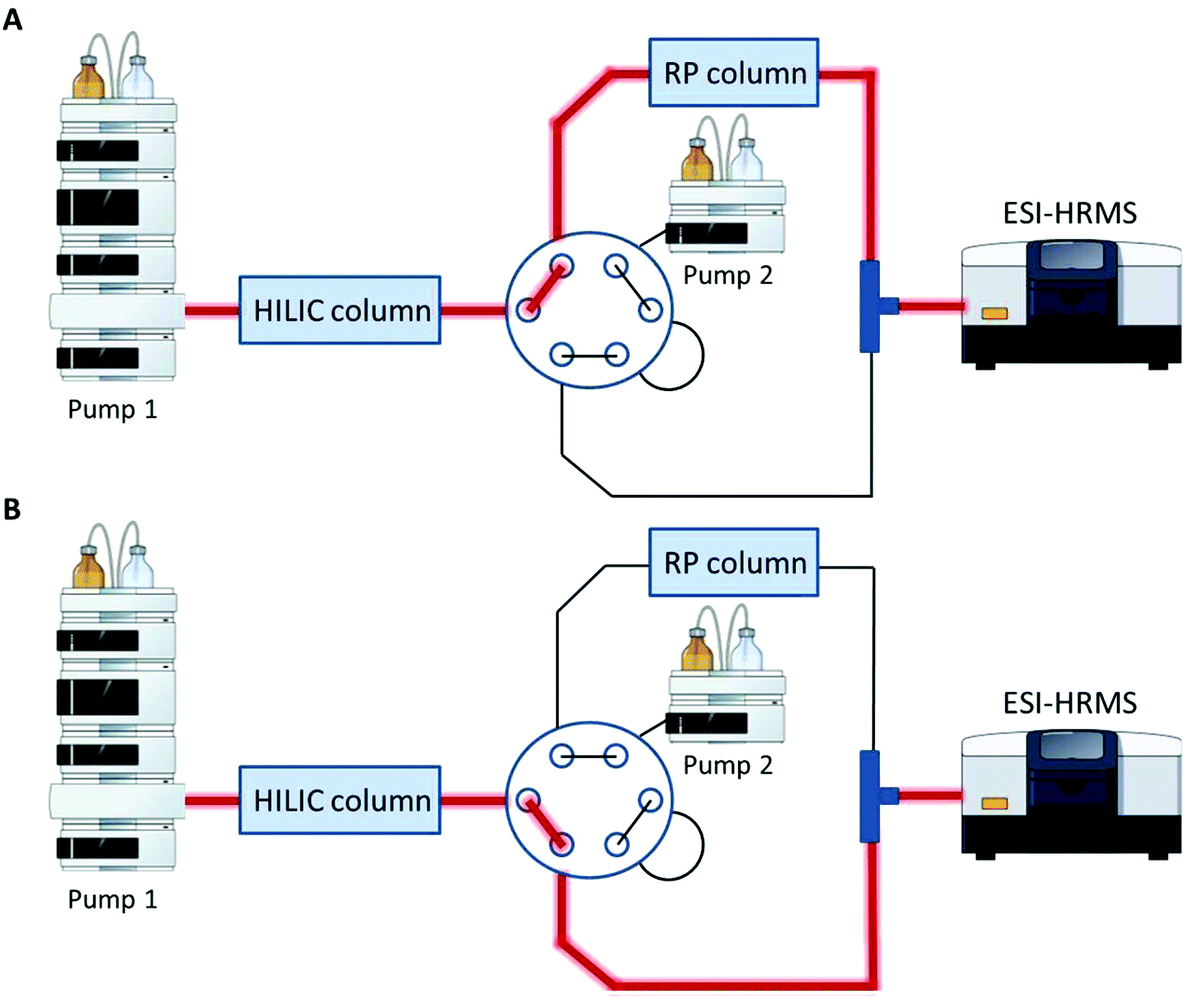
A Vanquish UHPLC (Thermo Fisher Scientific) equipped with an additional pump was used for HILIC and RP coupling. A previously published RP-PGC metabolite setup55 was adapted and further developed for lipid separation. The void volume including non-polar lipids was transferred from the first-dimension column (HILIC) to the second dimension (RP) using a separate two positional 6-port valve controlled by the mass spectrometer. The sample was diluted in 2 μL 95% ACN (and 5% H2O), injected onto the HILIC column and the HILIC and RP columns were serially coupled (position A, Fig. 1) from 0–1.5 min. All hydrophobic lipids not interacting with the HILIC column were transferred to the second RP dimension. At 1.5 min the valve was switched to position B (Fig. 1) directing the second-dimension pump flow to the RP column. The column effluents were combined using a T-piece prior to the introduction into the ESI source of the mass spectrometer.
 |
| | Fig. 1 Setup for HILIC-RP-MS. A two position six-port valve is used to transfer the void volume from the first dimension (HILIC) to the second dimension (RP). The valve was set to position A from 0–1.5 min, to position B from 1.5–14.1 min and for equilibration reasons prior to the next run again to position A from 14.1–15.0 min. A. Serial configuration: The HILIC column was directly connected with the RP column to transfer the void volume. B. Parallel configuration: The HILIC and the RP column used different eluent systems by two separate UHPLC pumps. Pump and MS icons were created via the Mind the Graph platform.56 | |
2.6 High-resolution mass spectrometry (HRMS)
High-resolution MS with a Q Exactive HF (Thermo Fisher Scientific) was used for lipid detection. The following HESI source parameters were applied: capillary temperature of 270 °C, sheath gas flow rate of 50, auxiliary flow rate of 14, sweep gas of 3, S-lens RF level of 45 and auxiliary gas heater temperature of 380 °C applying a spray voltage of 3.5 kV in positive mode and 2.8 kV in negative mode. Full-MS mode at 120000 resolution with an AGC target of 1e6 was used for the quantification runs. A top 10 ddMS2 method with inclusion list (using generated57 suspect lists for human plasma and yeast samples deduced from literature with assigned retention times determined by standards) was applied for the identification runs using 60000 MS1 resolution and 15000 MS2 resolution as well as normalized collision energies of 25 (+) and 28 (−). Spectral data was recorded in the mass range of 200–2000 m/z using profile mode. All triggered masses were set on the exclusion list for 15 s and if no masses of the inclusion list were found, ddMS2 spectra were recorded. Human plasma and yeast were also analyzed by direct infusion shotgun analysis using the robotic nanoflow ion source TriVersa NanoMate (Advion BioSciences, Ithaca NY, USA) into a Q Exactive HF instrument (Thermo Fisher Scientific, Bremen, Germany) followed by LipidXplorer analysis.11,58 Additional details are provided in the extended method section of the ESI.†
2.7 Data evaluation of HILIC-RP-HRMS
Data evaluation was performed using Lipid Search 4.1 (Thermo Fisher Scientific) for the ddMS2 identification runs (n = 3 samples and one sample was measured twice as analytical replicate, measurement in positive and negative mode). Lipid Search results were filtered for 5 ppm in MS1, 7 ppm in MS2 and the lipids were only considered if the areas were 3 × higher than in the blank samples or not present in the blanks at all. The main adduct ions in positive mode was set to H+ for PC, PS, PE, PA, HexCer, SM, AcCa, for MG, DG, TG, PG, PI, CE the main adduct ions were set to M + NH4/Na, for Cer, and HexCer additionally adduct ion with loss of H2O were considered. The main adduct ions in negative mode was set to H- for PS, PE, PA, Cer, HexCer, SM, AcCa, for PC and SM the main adduct ion was set to HCOO–, for Cer, and HexCer additionally adduct ion with loss of H2O were considered. The main grade was set to A (lipid class and fatty acids are completely identified) and B (lipid class and some fatty acids are identified) for all lipid classes except PC, Cer, HexCer and SM, there A, B and C (lipid class or fatty acids are identified) grade were allowed. Tracefinder 4.1 (Thermo Fisher Scientific) was used for Full-MS quantification of lipid standards based on peak areas obtained from extracted ion chromatograms (±5 ppm) with external calibration. The calibration was performed over four orders of magnitude (0.01–10 μM) for all lipids analysed. For lipid chromatograms Skyline (Version 3.7) was used.
3. Results and discussion
The chemical complexity of lipids demands robust and efficient chromatographic separations for profiling and quantification tasks in lipidomics. Here, we present a novel strategy for polar and non-polar lipid analysis based on an on-line combination of HILIC and RP chromatography to enable HILIC-RP-HRMS.
3.1 On-line coupled HILIC-RP liquid chromatography
Recently, an extensive comparative study of different HILIC columns for lipidomics application was performed.23 Based on these results, we chose an eluent system with 40 mM ammonium formate (pH = 4.0) and acetonitrile for HILIC chromatography. In this study, we used the Acquity BEH Amide (2.1 × 100 mm; 1.7 μm particle size, 130 Å pore size) enabling class-specific separation of polar lipids based on the hydrophilic head group. The starting conditions of 96% acetonitrile of the HILIC allowed to retain and separate HexCer (retention factor k > 2) from the void volume. However, a substantial lipid fraction still elutes in or near the void volume causing severe ion suppression in ESI-MS and subsequent problems with lipid identification and quantification. This fraction contains all non-polar lipids. As a consequence HILIC separations show poor performance for lipid classes such as TG, DG, MG, FA, ST, Cer or CE. Therefore, orthogonal chromatographic selectivity for lipid profiling of polar and non-polar lipids for a sample of interest is demanded. In this work, simultaneous polar and non-polar lipid analysis was achieved by combining the selectivity of HILIC and RP. For this purpose, a C18 column (Acquity UHPLC HSS T3) with sub-2 μm particle and UHPLC capabilities was chosen with a commonly used eluent system for lipids consisting of isopropanol, acetonitrile, water, ammonium formate and formic acid.14,22,27,34 The low-retaining non-polar lipid fraction (0–1.5 min) was directly transferred from the HILIC to the RP dimension (Fig. 1, 2 and Fig. S1†). This setup enables the on-line combination of HILIC with RP without any dilution (or use of a trapping column) in the second dimension. By exploiting hydrophobic and hydrophilic lipid interactions, (1) the use of a short HILIC column and direct transfer of the non-polar lipids without further dilution, (2) short gradient programs and (3) a relatively simple instrumental setup is enabled. The instrumental setup applied was adapted from a previously published RP-PGC metabolite method55 and further developed for lipid separation. In position A (Fig. 1A), the flow from the first UHPLC pump is used to separate polar lipids injected on the HILIC column. The RP column is coupled in a serial manner so that the non-polar lipids can interact with the apolar stationary phase. After the transfer of the whole void volume from the first to the second dimension, the valve is switched to the parallel configuration in position B (Fig. 1B). The polar lipids are eluted from the HILIC column within 6.5 min using an aqueous buffer gradient. At the same time, the second UHPLC pump is used to remove non-retained compounds from the RP column as well as the mobile phase originating from the HILIC dimension. After 7 min of chromatography time, non-polar lipid elution from the RP column was initiated using increasing isopropanol concentrations. Meanwhile, an 8 min equilibration step for the 100 mm HILIC column was carried out. After 9 min the RP column was switched back to starting conditions to allow 6 min of equilibration time for the RP column. This way, both the HILIC and the RP separation can be performed in 15 min including column reequilibration times. The effluents were united using a T-piece in front of the ESI-source prior to MS detection. Lipid standards from different classes (Cer, DAG, FA, HexCer, Hex2Cer, LPC, PA, PC, PE, PG, PI, PS, SM, ST, TG) were tested using the HILIC-RP setup. As can be seen in Fig. 2, ESI Fig. S1† and Table 1, successful transfer of the non-polar lipids to the second dimension was possible and enabled retention of polar and non-polar lipid with HILIC-RP.
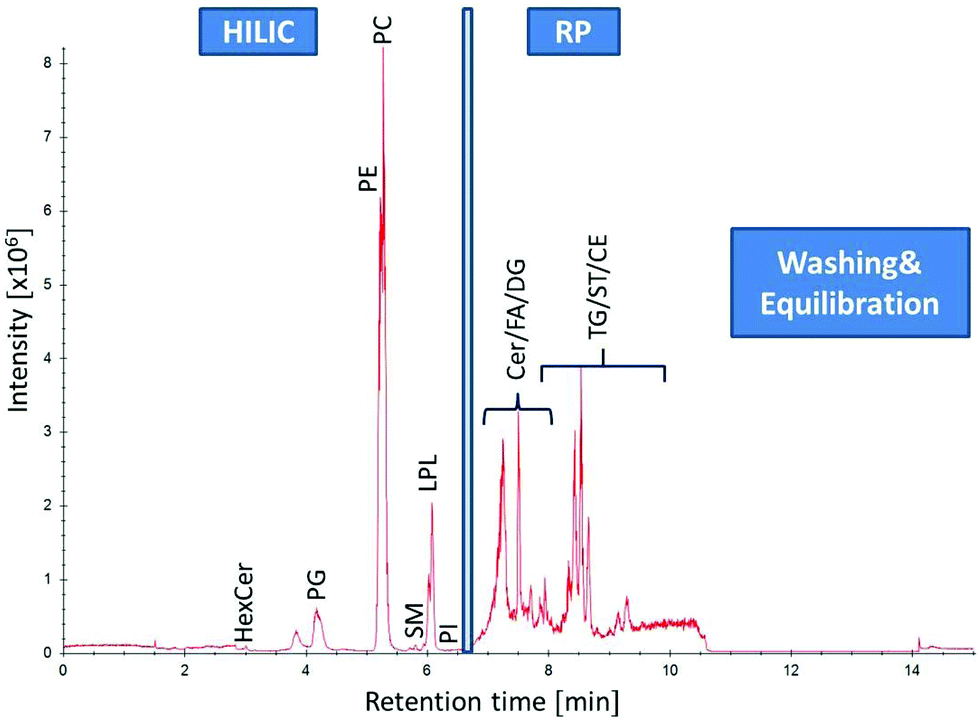
 |
| | Fig. 2 Separation of human plasma (SRM 1950) by HILIC-RP-HRMS. High resolution MS1 TIC in positive mode of different lipid classes: from 0–6.5 min lipid class separation of the HILIC column (Acquity UHPLC BEH Amide, 2.1 × 100 mm, 1.7 μm) is performed prior to RP (C18 Acquity UHPLC HSS T3, 2.1 mm × 150 mm, 1.8 μm) chromatography for non-polar lipid elution from 6.5–11 min followed by washing and equilibration step of both columns. | |
Table 1 Separation and quantification of 20 lipids originating from 15 lipid classes using RP-HILIC-HRMS (+/−) in human plasma (n = 3) and yeast (n = 3). Information on lipid short notation, MS polarity mode, retention time, linearity (R2), limit of detection (LOD), adduct formation, and m/z can be found as well as lipid concentrations determined in the samples can be found
| Lipid class |
Standards |
Sum formula |
MS |
RT (min) |
Linearity (R2) |
LOD (nM) |
Adduct |
m/z (+) |
m/z (−) |
SRM (n = 3) nmol mL−1 |
Yeast (n = 3) μmol 107 per cells |
| Average |
SD |
Average |
SD |
| Ceramide (Cer) |
Cer d36:1 |
C36H71NO3 |
+ |
7.88 |
0.9952 |
14 |
M + H |
566.551 |
|
0.18 |
0.01 |
0.011 |
0.004 |
| Cholesteryl ester (CE) |
CE 18:0 |
C45H80O2 |
+ |
9.65 |
0.9842 |
83 |
M + NH4 |
670.650 |
|
8.21 |
3.07 |
<LOD |
|
| Cholesteryl ester (CE) |
CE 18:2 |
C45H76O2 |
+ |
9.47 |
0.9959 |
83 |
M + NH4 |
666.618 |
|
73.33 |
18.93 |
0.006 |
0.001 |
| Diacylglycerol (DG) |
DG 34:1 |
C37H70O5 |
+ |
7.95 |
0.9933 |
5 |
M + NH4 |
617.512 |
|
8.90 |
0.66 |
0.651 |
0.085 |
| Fatty acid (FA) |
FA 16:0 |
C16H32O2 |
— |
7.48 |
0.9981 |
2 |
M − H |
|
255.233 |
64.43 |
3.52 |
4.493 |
0.662 |
| Fatty acid (FA) |
FA 18:0 |
C18H36O2 |
— |
7.64 |
0.9922 |
1 |
M − H |
|
283.265 |
9.58 |
0.05 |
4.237 |
0.836 |
| Hexosyl ceramide (HexCer) |
HexCer d34:1 |
C40H77NO8 |
+ |
3.01 |
0.9967 |
5 |
M + H |
700.572 |
|
<LOD |
|
<LOD |
|
| Dihexosylceramide (Hex2Cer) |
Hex2Cer d34:1 |
C46H87NO13 |
+ |
5.88 |
0.9970 |
59 |
M + H |
862.625 |
|
<LOD |
|
<LOD |
|
| Lysophosphatidylcholine (LPC) |
LPC 16:0 |
C24H50NO7P |
+/− |
6.1 |
0.9962 |
2 |
M + H/M + HCOO |
496.340 |
540.331 |
39.71 |
2.24 |
0.052 |
0.007 |
| Lysophosphatidylcholine (LPC) |
LPC 18:0 |
C26H54NO7P |
+/− |
6.03 |
0.9965 |
1 |
M + H/M + HCOO |
524.371 |
568.362 |
23.93 |
2.13 |
0.029 |
0.004 |
| Phosphatidic acid (PA) |
PA 36:2 |
C39H73O8P |
— |
5.62 |
0.9933 |
1015 |
M − H |
|
699.497 |
<LOD |
|
19.616 |
3.272 |
| Phosphatidylcholine (PC) |
PC 34:1 |
C42H82NO8P |
+/− |
5.3 |
0.9921 |
1 |
M + H/M + HCOO |
760.585 |
804.576 |
65.09 |
3.99 |
2.501 |
0.434 |
| Phosphatidylcholine (PC) |
PC 34:2 |
C42H80NO8P |
+/− |
5.31 |
0.9916 |
1 |
M + H/M + HCOO |
758.569 |
802.560 |
122.62 |
7.02 |
10.678 |
1.558 |
| Phosphatidylcholine (PC) |
PC O-36:2 |
C44H86NO7P |
+ |
5.23 |
0.9965 |
1 |
M + H/M + Cl |
772.621 |
806.584 |
0.78 |
0.60 |
4.139 |
0.841 |
| Phosphatidylethanolamine (PE) |
PE 36:2 |
C41H78NO8P |
+/− |
5.31 |
0.9940 |
4 |
M + H/M − H |
744.554 |
742.539 |
5.15 |
0.16 |
39.125 |
6.291 |
| Phosphatidylglycerol (PG) |
PG 36:2 |
C42H79O10P |
+/− |
4.26 |
0.9955 |
76 |
M + H/M − H |
775.548 |
773.534 |
<LOD |
|
0.078 |
0.009 |
| Phosphatidylserine (PS) |
PS 36:2 |
C42H78NO10P |
— |
6.19 |
0.9889 |
640 |
M − H |
|
786.529 |
<LOD |
|
3.310 |
0.790 |
| Sphingomyelin (SM) |
SM d42:2 |
C47H93N2O6P |
+ |
5.71 |
0.9858 |
6 |
M + H |
813.684 |
|
10.20 |
0.83 |
<LOD |
|
| Sterol (ST) |
ST 28:3 |
C28H44O |
+ |
7.85 |
0.9568 |
104 |
M + H |
397.347 |
|
<LOD |
|
2.981 |
0.403 |
| Triacylglycerol (TG) |
TG 52:2 |
C55H102O6 |
+ |
8.67 |
0.9931 |
4 |
M + NH4 |
881.757 |
|
7.10 |
1.36 |
0.564 |
0.047 |
Compared to the one-dimensional HILIC and RP lipid separations, no band broadening was introduced (Full width at half maximum/FWHM of 4–6 s for one-dimensional RP, HILIC and the HILIC-RP coupling) by the suggested instrumental setup despite mixing of the mobile phases from the two columns was performed (Fig. 3).
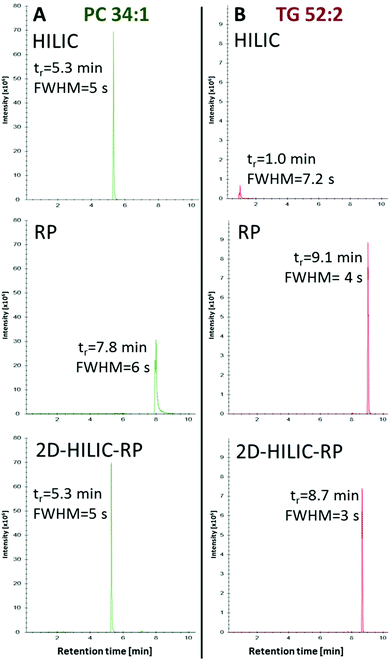 |
| | Fig. 3 Comparison of peak widths using HILIC and RP separately or coupled (HILIC-RP), shown exemplarily for the extracted ion chromatograms of PC 34:1 (A) and TG 52:2 (B) detected by HRMS in positive mode. Retention time (tr) is given in minutes and the peak width is calculated via full peak width at half-maximal peak height (FWHM). The chromatogram shows that the non-polar lipids were successfully loaded onto the RP column while the comparison between HILIC and HILIC-RP peak widths shows that the on-line coupling does not introduce peak broadening. | |
Using this novel HILIC-RP-HRMS method, high-throughput polar and non-polar lipid analysis is possible within 15 min enabling class-specific HILIC separation lipids without the loss of the non-polar lipid fraction observed for conventional HILIC methods.16,23,28,30–32 The fast chromatographic run time of 15 min is comparable to state of the art direct-infusion shotgun lipidomics profiling approaches.10–12
3.2 HILIC-RP-HRMS applications in lipidomics
After successful method development, the final HILIC-RP-MS method was applied to characterize two different sample types namely Pichia pastoris yeast and human serum (SRM 1950) samples. Lipids were extracted accordingly by Folch extraction53 for yeast samples (n = 3) and MTBE extraction for human plasma samples (n = 3).54 On-line HILIC-RP chromatography was coupled to a high-resolution mass spectrometer (Q Exactive HF).
3.3 Untargeted screening by HILIC-RP-HRMS
A panel of 20 lipid standards covering 14 lipid classes (CE, DG, HexCer, Hex2Cer, FA, LPC, PA, PC, PE, PG, PS, SM, ST, TG) was used to test correct annotations based on accurate m/z values (mass accuracy <5 ppm), characteristic fragmentation in both polarity modes and retention time behaviour (Table 1). All tested standards were unambiguously identified and class-specific coelution on the HILIC column was observed for all annotated polar lipids (Table 1, ESI Table S1†). Untargeted lipid screening of human plasma resulted in ∼ 400 lipid annotations from 14 different lipid classes (Cer, HexCer, CE, MG, DG, FA, LPC, LPE, PC, PE, PI, SM, AcCa, TG) (ESI Table S1†). Untargeted profiling of yeast samples led to the identification of ∼130 lipids from 14 different lipid classes (DG, LPC, LPE, PC, PE, MG, HexCer, DMPE, PG, PI, PS, TG, Cer, Co) (ESI Table S1†). A summary of all lipids identified by MS2 based annotations in human plasma and yeast samples can be found in the ESI (Table S1†). It has to be noted that the number of lipid annotations is dependent on the level of structural information determined by the analytical method i.e. using the HILIC-RP-HRMS method the lipid class can be determined by chromatographic retention time, the molecular lipid species by accurate m/z information (MS1) and the fatty acyl/alkyl level by fragmentation (MS2). Overall 58% of the MS2 annotated lipids in both sample types showed retention on the HILIC dimension (2–6.5 min) and 42% of lipids were eluted from the second RP dimension (7–10 min). Comparing HILIC-RP-HRMS to other lipidomics workflows, the characteristics are similar to classical shotgun analysis in terms of run time (15 min) and the presence of intra-class lipid competition for ionization. To test the benefit of the chromatographic dimension to prevent co-elution of isobars from different lipid classes, we compared profiling results from human plasma and yeast samples received by HILIC-RP-HRMS and shotgun MS (ESI Table S1†). The number of lipid species level annotations (human plasma SRM 1950: ∼400; Pichia pastoris yeast: ∼130) achieved using HILIC-RP-HRMS were significantly higher compared to shotgun MS (human plasma SRM 1950: ∼200, Pichia pastoris yeast: ∼90) (ESI Tables S1 and S2†). These results are consistent with literature reports on human plasma SRM 1950 analysis, where 250 lipids were identified with shotgun MS compared to 520 lipid annotations by a 1.5 h RP-MS workflow.59,60 HILIC-RP-MS analysis enabled the identification of additional lipid classes such as AcCa, DMPE and higher lipid annotation numbers for low abundant classes such as Cer, HexCer, Co compared to shotgun MS (ESI Tables S1 and S2†). In shotgun MS higher abundance of lysophospholipids (ESI Tables S1 and S2†) and fatty acids (data not shown) species was observed. This highlights one general problem of shotgun MS to differentiate fatty acids and lysophospholipids present in the sample and in-source fragments produced in the MS. HILIC-RP-MS on the other hand, can easily resolve lipids from in-source fragments using the additional retention time information.
Assessment of HILIC-RP-HRMS quantification capabilities.
Finally, the novel HILIC-RP-HRMS approach was investigated regarding the analytical figures of merit of retention time stability, linear dynamic range and limit of detection using a panel of 20 lipids (Table 1). Linear calibration curves over 4 orders of magnitude could be obtained for most standards (0.01–10 μM) (Table 1, ESI Fig. S3†). LODs were calculated using the lowest detected concentration point (n = 3) of the calibration61 and were found in the low to high nM range (corresponding to low fmol to low pmol absolute on column) depending on the lipid species analyzed (Table 1) as reported for other LC-MS based lipidomics approaches.62,63 Compared to classical shotgun based workflows, the method was superior, as previous results reported LODs and LOQs in the low to high μM range for the target lipids.64 Both the retention time and the peak area precision were excellent with RSDs ≤ 1% and <5% respectively (over a measurement period of 44 h) for all lipids monitored. In a next step, preliminary quantification of 20 target lipid was performed by external calibration in SRM 1950 human plasma (n = 3) and Pichia pastoris yeast (n = 3) samples. Lipid concentrations determined in the latter sample type were in the nmol to μmol range for 107 cells (Table 1) and compared well to previous studies on Pichia pastoris or other yeasts.34,65–68 Trueness bias of analysis was assessed in the SRM 1950 using the recently introduced accuracy assessment tool LipidQC,69 which followed the publication of a NIST interlaboratory comparison report on high molecular lipids70,71 (ESI Fig. S4†). After normalization by volume the obtained values proved to be in agreement with the reported values for several lipid classes as all quantified lipids were in the nmol mL−1 range. It has to be noted however, that accurate absolute quantification strategies in lipidomics involve internal standardization. External calibration without internal standardization as carried out in the proof of principle experiments here is not recommended for lipids retained on reversed phase separations. Despite this fact, the preliminary data on the reference material exemplified the potential of the HILIC-RP-HRMS method for future quantification studies (involving internal standards). As can be seen in ESI Fig. S4,† the accuracy assessment revealed that 13 out of 15 lipid standards (FA 16:0 DG 34:1, TG 52:2, LPC 16:0, LPC 18:0, PC 34:1, PC 34:2, PC O-36:2, PE 36:2, Cer d36:1, SM d42:2, CE 18:0, CE 18:2) fell within the 99% confidence interval of the consensus mean value,69 despite the fact that only external calibration was implemented not compensating for matrix effects and losses during sample preparation. This preliminary data show the potential of the HILIC-RP-HRMS method. Future studies will focus on implementation of a routine quantification method establishing internal standards and automatic deisotoping algorithm. Overall, the results show that fast in-depth profiling and quantification is possible by HILIC-RP-HRMS with increased level of lipid identification by retention time information. The class-specific separation on the HILIC column allows class-specific lipid quantification in 15 min as performed in state of the art shotgun lipidomics approaches.10,72,73 Moreover, the on-line coupled RP column enables additional non-polar lipid trapping compared to conventional HILIC methods.28 Finally, co-elution of isobars from different lipid classes is avoided and the linear dynamic range is increased compared to shotgun lipidomics approaches. Hence, HILIC-RP-MS is able to combine several interesting features (1) the decreased run time of state of the art shotgun MS methods, (2) the elevated linear dynamic range inherent to chromatographic separation and (3) increased level of identification by separation of polar and non-polar lipid classes.
4. Conclusion
With the advent of sub-2 μm particles and UHPLC new chromatographic possibilities became available.37–41 In this work, we show the power of on-line combinations of sub-2 μm HILIC and RP coupled to HRMS for lipid analysis. By addition of a 6-port valve and a T-piece, direct coupling of the HILIC column to the RP column was possible for simultaneous analysis of polar and non-polar lipids. This setup enables on-line combination of HILIC with RP without any dilution (or use of a trapping column) in the second dimension, exploiting the strong interaction of hydrophobic lipids and the enhanced separation space offered by the orthogonal methods. The fast chromatographic run time of 15 min is comparable to state of the art direct-infusion shotgun lipidomics profiling approaches.10–12 These results show that HILIC-RP-HRMS is a valuable tool for high-throughput lipidomics analysis, bridging the gap between state of the art shotgun and LC-MS approaches. Moreover, we strongly believe that lipidomics studies will benefit from the increased separation space, enhanced sample throughput and broader lipid information of on-line coupled HILIC-RP-MS methods.
Abbreviations
| ST | Sterols |
| GPL | Glycerophospholipids |
| DG | Diglyceride |
| TG | Triglyceride |
| MG | Monoglyceride |
| Cer | Ceramide |
| CE | Cholesterol ester |
| PE | Phosphatidylethanolamine |
| LPE | Lysophosphatidylethanolamine |
| DMPE | Dimethyl-phosphatidylethanolamine |
| PC | Phosphatidylcholine |
| LPC | Lysophosphatidylcholine |
| PG | Phosphatidylglycerol |
| PI | Phosphatidylinositol |
| PS | Phosphatidylserine |
| PA | Phosphatidic acid |
| SM | Sphingomyelin |
| FA | Fatty acids |
| AcCa | Acyl carnitine |
| HexCer | Hexosyl ceramide |
| Hex2Cer | Dihexosyl ceramide |
| Co | Coenzyme |
| HRMS | High resolution mass spectrometry |
| HILIC | Hydrophilic interaction chromatography |
| RP | Reversed-phase chromatography |
| UHPLC | Ultra-high performance liquid chromatography |
Conflicts of interest
The authors declare no competing financial interest.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version.
Acknowledgements
This work was supported by the University of Vienna, the Faculty of Chemistry, the Vienna Metabolomics Center (VIME; http://metabolomics.univie.ac.at/) and the research platform Chemistry Meets Microbiology of the University of Vienna. The authors thank all members of the Environmental Analysis (University of Vienna) group for continuous support. Especially, we acknowledge Petra Volejnik and Sophie Neumayer for laboratory support. Moreover, the collaborative work with Gerrit Hermann (ISOtopic Solutions, Vienna) is highly appreciated.
References
- E. Fahy, J. Lipid Res., 2005, 46, 839–862 CrossRef CAS PubMed.
-
X. Han, Lipidomics: Comprehensive mass spectrometry of lipids, John Wiley & Sons, Hoboken, New Jersey, 2016 Search PubMed.
- M. Sud, E. Fahy, D. Cotter, A. Brown, E. A. Dennis, C. K. Glass, A. H. Merrill, R. C. Murphy, C. R. H. Raetz, D. W. Russell and S. Subramaniam, Nucleic Acids Res., 2007, 35, 527–532 CrossRef PubMed.
-
http://www.lipidmaps.org
.
- E. Fahy, S. Subramaniam, R. C. Murphy, M. Nishijima, C. R. H. Raetz, T. Shimizu, F. Spener, G. van Meer, M. J. O. Wakelam and E. a. Dennis, J. Lipid Res., 2009, 50(Suppl), S9–S14 CrossRef PubMed.
- G. van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112–124 CrossRef CAS PubMed.
- S. Daemen, M. A. M. J. van Zandvoort, S. H. Parekh and M. K. C. Hesselink, Mol. Metab., 2016, 5, 153–163 CrossRef CAS PubMed.
- S. Martin and R. G. Parton, Semin. Cell Dev. Biol., 2005, 16, 163–174 CrossRef CAS PubMed.
- A. Shevchenko and K. Simons, Nat. Rev. Mol. Cell Biol., 2010, 11, 593–598 CrossRef CAS PubMed.
- D. Schwudke, K. Schuhmann, R. Herzog, S. Bornstein and A. Shevchenko, Cold Spring Harbor Perspect. Biol., 2011, 3, 1–13 Search PubMed.
- R. Herzog, K. Schuhmann, D. Schwudke, J. L. Sampaio, S. R. Bornstein, M. Schroeder and A. Shevchenko, PLoS One, 2012, 7, 15–20 Search PubMed.
- A. Shevchenko and K. Simons, Nat. Rev. Mol. Cell Biol., 2010, 11, 593–598 CrossRef CAS PubMed.
- P. Rainville and C. Stumpf, J. Proteome Res., 2007, 6, 552–558 CrossRef CAS PubMed.
- A. Fauland, H. Kofeler, M. Trotzmuller, A. Knopf, J. Hartler, A. Eberl, C. Chitraju, E. Lankmayr and F. Spener, J. Lipid Res., 2011, 52, 2314–2322 CrossRef CAS PubMed.
- S. S. Bird, V. R. Marur, M. J. Sniatynski, H. K. Greenberg and B. S. Kristal, Anal. Chem., 2011, 83, 6648–6657 CrossRef CAS PubMed.
- E. Cifkova, M. Holcapek, M. Lisa, M. Ovcacikova, A. Lycka, F. Lynen and P. Sandra, Anal. Chem., 2012, 84, 10064–10070 CrossRef CAS PubMed.
- J. L. Desantos-Garcia, S. I. Khalil, A. Hussein, Y. Hu and Y. Mechref, Electrophoresis, 2011, 32, 3516–3525 CrossRef CAS PubMed.
- K. Sandra and P. Sandra, Curr. Opin. Chem. Biol., 2013, 17, 847–853 CrossRef CAS PubMed.
- M. Smith and F. B. Jungalwala, J. Lipid Res., 1981, 22, 697–704 CAS.
- J. J. Myher and A. Kuksis, J. Chromatogr. B, 1995, 5, 3-33–33 Search PubMed.
- D. Y. Bang and M. H. Moon, J. Chromatogr. A, 2013, 1310, 82–90 CrossRef CAS PubMed.
- O. L. Knittelfelder, B. P. Weberhofer, T. O. Eichmann, S. D. Kohlwein and G. N. Rechberger, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 951–952, 119–128 CrossRef CAS PubMed.
- E. Cífková, R. Hájek, M. Lísa and M. Holcapek, J. Chromatogr. A, 2016, 1439, 65–73 CrossRef PubMed.
- M. Hermansson, A. Uphoff, R. Käkelä and P. Somerharju, Anal. Chem., 2005, 77, 2166–2175 CrossRef CAS PubMed.
- M. Lísa and M. Holčapek, Anal. Chem., 2015, 87, 7187–7195 CrossRef PubMed.
- P. T.-S. Pei, R. S. Henly and S. Ramachandran, Lipids, 1975, 10, 152–156 CrossRef CAS PubMed.
- B. Peng and R. Ahrends, J. Proteome Res., 2016, 15, 291–301 CrossRef CAS PubMed.
- M. Lísa, E. Cífková and M. Holčapek, J. Chromatogr. A, 2011, 1218, 5146–5156 CrossRef PubMed.
- A. Fauland, H. Köfeler, M. Trötzmüller, A. Knopf, J. Hartler, A. Eberl, C. Chitraju, E. Lankmayr and F. Spener, J. Lipid Res., 2011, 52, 2314–2322 CrossRef CAS PubMed.
- C. Zhu, A. Dane, G. Spijksma, M. Wang, J. van der Greef, G. Luo, T. Hankemeier and R. J. Vreeken, J. Chromatogr. A, 2012, 1220, 26–34 CrossRef CAS PubMed.
- Y. Okazaki, Y. Kamide, M. Y. Hirai and K. Saito, Metabolomics, 2013, 9, 121–131 CrossRef CAS PubMed.
-
G. Vanhoenacker, R. Kindt, F. David, P. Sandra and K. Sandra, Agilent Application Note, 2015, 5991–5532EN, https://www.agilent.com/cs/library/applications/5991-5532EN.pdf Search PubMed.
- P. Dugo, O. Favoino, R. Luppino, G. Dugo and L. Mondello, Anal. Chem., 2004, 76, 2525–2530 CrossRef CAS PubMed.
- E. Rampler, C. Coman, G. Hermann, A. Sickmann, R. Ahrends and G. Koellensperger, Analyst, 2017, 142, 1891–1899 RSC.
- X. Gao, Q. Zhang, D. Meng, G. Isaac, R. Zhao, T. L. Fillmore, R. K. Chu, J. Zhou, K. Tang, Z. Hu, R. J. Moore, R. D. Smith, M. G. Katze and T. O. Metz, Anal. Bioanal. Chem., 2012, 402, 2923–2933 CrossRef CAS PubMed.
- S. Granafei, P. Azzone, V. A. Spinelli, I. Losito, F. Palmisano and T. R. I. Cataldi, J. Chromatogr. A, 2016, 1477, 47–55 CrossRef CAS PubMed.
- D. T.-T. Nguyen, D. Guillarme, S. Rudaz and J.-L. Veuthey, J. Sep. Sci., 2006, 29, 1836–1848 CrossRef CAS PubMed.
- R. Hayes, A. Ahmed, T. Edge and H. Zhang, J. Chromatogr. A, 2014, 1357, 36–52 CrossRef CAS PubMed.
- T. L. Chester, Anal. Chem., 2013, 85, 579–589 CrossRef CAS PubMed.
- M. Witting, T. V. Maier, S. Garvis and P. Schmitt-Kopplin, J. Chromatogr. A, 2014, 1359, 91–99 CrossRef CAS PubMed.
- J. O. Omamogho and J. D. Glennon, Anal. Chem., 2011, 83, 1547–1556 CrossRef CAS PubMed.
- M. Holčapek, M. Ovčačíková, M. Lísa, E. Cífková and T. Hájek, Anal. Bioanal. Chem., 2015, 407, 5033–5043 CrossRef PubMed.
- M. Li, B. Feng, Y. Liang, W. Zhang, Y. Bai, W. Tang, T. Wang and H. Liu, Anal. Bioanal. Chem., 2013, 405, 6629–6638 CrossRef CAS PubMed.
- P. Jandera, T. Hájek, M. Staňková, K. Vyňuchalová and P. Česla, J. Chromatogr. A, 2012, 1268, 91–101 CrossRef CAS PubMed.
- P. Česla and J. Křenková, J. Sep. Sci., 2017, 40, 109–123 CrossRef PubMed.
- D. R. Stoll, X. Li, X. Wang, P. W. Carr, S. E. G. Porter and S. C. Rutan, J. Chromatogr. A, 2007, 1168, 3–2 CrossRef CAS PubMed.
- C. R. Evans and J. W. Jorgenson, Anal. Bioanal. Chem., 2004, 378, 1952–1961 CrossRef CAS PubMed.
- S. S. Brudin, R. A. Shellie, P. R. Haddad and P. J. Schoenmakers, J. Chromatogr. A, 2010, 1217, 6742–6746 CrossRef CAS PubMed.
- P. Q. Tranchida, P. Donato, G. Dugo, L. Mondello and P. Dugo, TrAC, Trends Anal. Chem., 2007, 26, 191–205 CrossRef CAS.
- P. Dugo, F. Cacciola, T. Kumm, G. Dugo and L. Mondello, J. Chromatogr. A, 2008, 1184, 353–368 CrossRef CAS PubMed.
- A. L. Huidobro, P. Pruim, P. Schoenmakers and C. Barbas, J. Chromatogr. A, 2008, 1190, 182–190 CrossRef CAS PubMed.
- S. Neubauer, C. Haberhauer-Troyer, K. Klavins, H. Russmayer, M. G. Steiger, B. Gasser, M. Sauer, D. Mattanovich, S. Hann and G. Koellensperger, J. Sep. Sci., 2012, 35, 3091–3105 CrossRef CAS PubMed.
- J. Folch, M. Lees and G. H. Sloane Stanley, J. Biol. Chem., 1952, 226, 497–509 Search PubMed.
- V. Matyash, G. Liebisch, T. V. Kurzchalia, A. Shevchenko and D. Schwudke, J. Lipid Res., 2008, 49, 1137–1146 CrossRef CAS PubMed.
- K. Ortmayr, S. Hann and G. Koellensperger, Analyst, 2015, 140, 3465–3473 RSC.
-
https://mindthegraph.com/
.
- P. Husen, K. Tarasov, M. Katafiasz, E. Sokol, J. Vogt, J. Baumgart, R. Nitsch, K. Ekroos and C. S. Ejsing, PLoS One, 2013, 8(11), e79736 CAS.
- C. Coman, F. A. Solari, A. Hentschel, A. Sickmann, R. P. Zahedi and R. Ahrends, Mol. Cell. Proteomics, 2016, 15, 1453–1466 CAS.
- T. Kind, K. H. Liu, D. Y. Lee, B. Defelice, J. K. Meissen and O. Fiehn, Nat. Methods, 2013, 10, 755–758 CrossRef CAS PubMed.
- K. Sandra, A. dos S. Pereira, G. Vanhoenacker, F. David and P. Sandra, J. Chromatogr. A, 2010, 1217, 4087–4099 CrossRef CAS PubMed.
-
B. Magnusson and U. Örnemark, Eurachem Guide: The Fitness for Purpose of Analytical Methods – A Laboratory Guide to Method Validation and Related Topics, 2nd edn, 2014 Search PubMed.
- X. Liu, Z. Ser and J. W. Locasale, Anal. Chem., 2014, 86, 2175–2184 CrossRef CAS PubMed.
- J. Zhou, H. Liu, Y. Liu, J. Liu, X. Zhao and Y. Yin, Anal. Chem., 2016, 88, 4478–4486 CrossRef CAS PubMed.
- M. A. Surma, R. Herzog, A. Vasilj, C. Klose, N. Christinat, D. Morin-Rivron, K. Simons, M. Masoodi and J. L. Sampaio, Eur. J. Lipid Sci. Technol., 2015, 117, 1540–1549 CrossRef CAS PubMed.
- C. S. Ejsing, J. L. Sampaio, V. Surendranath, E. Duchoslav, K. Ekroos, R. W. Klemm, K. Simons and A. Shevchenko, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2136–2141 CrossRef CAS PubMed.
- L. Klug, P. Tarazona, C. Gruber, K. Grillitsch, B. Gasser, M. Trötzmüller, H. Köfeler, E. Leitner, I. Feussner, D. Mattanovich, F. Altmann and G. Daum, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2014, 1841, 215–226 CrossRef CAS PubMed.
- V. A. Ivashov, K. Grillitsch, H. Koefeler, E. Leitner, D. Baeumlisberger, M. Karas and G. Daum, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2013, 1831, 282–290 CrossRef CAS PubMed.
- K. Grillitsch, P. Tarazona, L. Klug, T. Wriessnegger, G. Zellnig, E. Leitner, I. Feussner and G. Daum, Biochim. Biophys. Acta, 2014, 1838, 1889–1897 CrossRef CAS PubMed.
- C. Z. Ulmer, J. M. Ragland, J. P. Koelmel, A. Heckert, C. M. Jones, T. Garrett, R. A. Yost and J. A. Bowden, Anal. Chem., 2017, 89(24), 13069–13073 CrossRef CAS PubMed.
-
J. A. Bowden, C. Z. Ulmer, C. M. Jones and A. Heckert, National Institute of Standards and Technology-Interagency/Internal Report (NISTIR), 2017, vol. 8185, pp. 1–451 Search PubMed.
- J. A. Bowden, A. Heckert, C. Z. Ulmer, C. M. Jones, J. P. Koelmel, L. Abdullah, L. Ahonen, Y. Alnouti, A. Armando, J. M. Asara, T. Bamba, J. R. Barr, J. Bergquist, C. H. Borchers, J. Brandsma, S. B. Breitkopf, T. Cajka, A. Cazenave-Gassiot, A. Checa, M. A. Cinel, R. A. Colas, S. Cremers, E. A. Dennis, J. E. Evans, A. Fauland, O. Fiehn, M. S. Gardner, T. J. Garrett, K. H. Gotlinger, J. Han, Y. Huang, A. Huipeng Neo, T. Hyötyläinen, Y. Izumi, H. Jiang, H. Jiang, J. Jiang, M. Kachmann, R. Kiyonami, K. Klavins, C. Klose, H. C. Köfeler, J. Kolmert, T. Koal, G. Koster, Z. Kuklenyik, I. J. Kurland, M. Leadley, K. Lin, K. R. Maddipati, M. Danielle, P. J. Meikle, N. A. Mellett, C. Monnin, M. A. Moseley, R. Nandakumar, M. Oresic, R. Patterson, D. Peake, J. S. Pierce, M. Post, A. D. Postle, R. Pugh, Y. Qiu, O. Quehenberger, P. Ramrup, J. Rees, B. Rembiesa, D. Reynaud, M. R. Roth, S. Sales, K. Schuhmann, M. L. Schwartzman, C. N. Serhan, A. Shevchenko, S. E. Sommerville, L. St. John-Williams, M. A. Surma, H. Takeda, R. Thakare, J. W. Thompson, F. Torta, A. Triebl, M. Trötzmüller, K. Ubhayasekera, D. Vuckovic, J. M. Weir, R. Welti, M. R. Wenk, C. E. Wheelock, L. Yao, M. Yuan, X. H. Zhao and S. Zhou, J. Lipid Res., 2017, 58(12), 2275–2288 CrossRef CAS PubMed.
- C. S. Ejsing, J. L. Sampaio, V. Surendranath, E. Duchoslav, K. Ekroos, R. W. Klemm, K. Simons and A. Shevchenko, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2136–2141 CrossRef CAS PubMed.
- K. Yang and X. Han, Metabolites, 2011, 1, 21–40 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an01984j |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *abc,
Harald
Schoeny
*abc,
Harald
Schoeny