
Expanding the structural analysis capabilities on an Orbitrap-based mass spectrometer for large macromolecular complexes†
Kyle L.
Fort‡
abc,
Michiel
van de Waterbeemd‡
ab,
Dmitriy
Boll
c,
Maria
Reinhardt-Szyba
c,
Mikhail E.
Belov
c,
Eita
Sasaki
d,
Reinhard
Zschoche
d,
Donald
Hilvert
d,
Alexander A.
Makarov
ac and
Albert J. R.
Heck
 *ab
*ab
aBiomolecular Mass Spectrometry and Proteomics, Bijvoet Center for Biomolecular Research and Utrecht Institute of Pharmaceutical Sciences, Utrecht University, 3584 Utrecht, The Netherlands. E-mail: a.j.r.heck@uu.nl
bNetherlands Proteomics Center, 3584 Utrecht, The Netherlands
cThermo Fisher Scientific (Bremen), 28199 Bremen, Germany
dLaboratory of Organic Chemistry, ETH Zürich, 8093 Zürich, Switzerland
First published on 8th November 2017
Abstract
Native mass spectrometry can provide insight into the structure of macromolecular biological systems. As analytes under investigation get larger and more complex, instrument capabilities need to be advanced. Herein, modifications to an Orbitrap Q Exactive Plus mass spectrometer that increase signal intensity, mass resolution, and maximum m/z measurable are described.
The application of mass spectrometry (MS) has undergone a paradigm shift from the analysis of small organic molecules to that of intact biomolecular assemblies. While early mass spectrometric investigations of biological systems were limited by the then-available ionization processes, the development of modern ionization methods, in particular nano-electrospray ionization (nESI), have overcome this limitation.1–3 Since then, technological advancements in MS instrumentation and additional developments in sample preparation have been the driving force in increasing the size of the analyte able to be investigated and the depth of structural detail that can be elucidated.4–10 With these advancements, the field of native MS has emerged, demonstrating that when proteins are subjected to nESI in the presence of buffering conditions, their native fold can be partially retained in the gas phase and analyzed with MS, revealing details with regard to the protein molecular weight, the complex stoichiometry, and protein ligand binding.11,12
While the analysis of these high molecular weight systems has been successfully performed on time-of-flight mass spectrometers, more recent developments have demonstrated that Orbitrap™-based mass spectrometers can be modified to perform these analyses, with the ability to transmit and detect ion signals for biomolecular assemblies of up to MDa in size.8,13 However, although commercially available Orbitrap mass spectrometers have performed well for analyses of large single proteins and small protein complexes, further advances in instrumentation are required to advance to the routine analysis of increasingly large biological systems, e.g., intact ribosomes, membrane ion channels, and virus capsids. Currently, Orbitrap instruments can detect ion signals up to about 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m/z; however, at these values, the transmission of these ions is low and prohibits the use of long transient length acquisitions, which are typically needed for optimal mass resolution. Thus, to enhance the information obtainable with native MS, the ion transmission properties of high (>20000) m/z ions must be increased.
000 m/z; however, at these values, the transmission of these ions is low and prohibits the use of long transient length acquisitions, which are typically needed for optimal mass resolution. Thus, to enhance the information obtainable with native MS, the ion transmission properties of high (>20000) m/z ions must be increased.
Herein, we sought to make modifications to the Orbitrap platform to further increase the sensitivity and transmission properties for very large protein complexes. Collectively, these modifications greatly increase the transmission and detection of ions at very high m/z values on the Q Exactive Plus mass spectrometer, whereby the abundance of the signal is enhanced 5–10 fold and ion signals up to 70000 m/z can be detected. Preliminary data acquired with the modifications described here have already been reported through the analysis of E. coli ribosomal particles.14 Here, we provide more in-depth details on all the modifications made, and describe much more technical data on the performance characteristics of the new instrument.
Methods and materials
Instrument modification
The exterior electronic boards of the mass spectrometer were modified to allow for the lowering of the RF ion optic frequencies, as well as allowing for pulsing of the SRIG exit lens, the DC bias of the injection flatapole, and the injection flatapole lens (Fig. 1a), allowing for in-source trapping. The details of in-source trapping are described in detail below (see section: Implementation of in-source trapping). The RF frequencies on the bent flatapole, quadrupole, transfer multipole, C-trap, and HCD-cell were modified to be 1.1 MHz, 287 kHz, 900 kHz, 2.1 MHz, and 900 kHz, respectively. The modification of these frequencies was aimed at increasing the transmission at high m/z, and their collective effect is described below. Additionally, the HCD-cell collision energy (CE) was increased to 300 V and the voltage ramp time of the Orbitrap deflector was adjusted to allow longer fill times of the mass analyzer cell.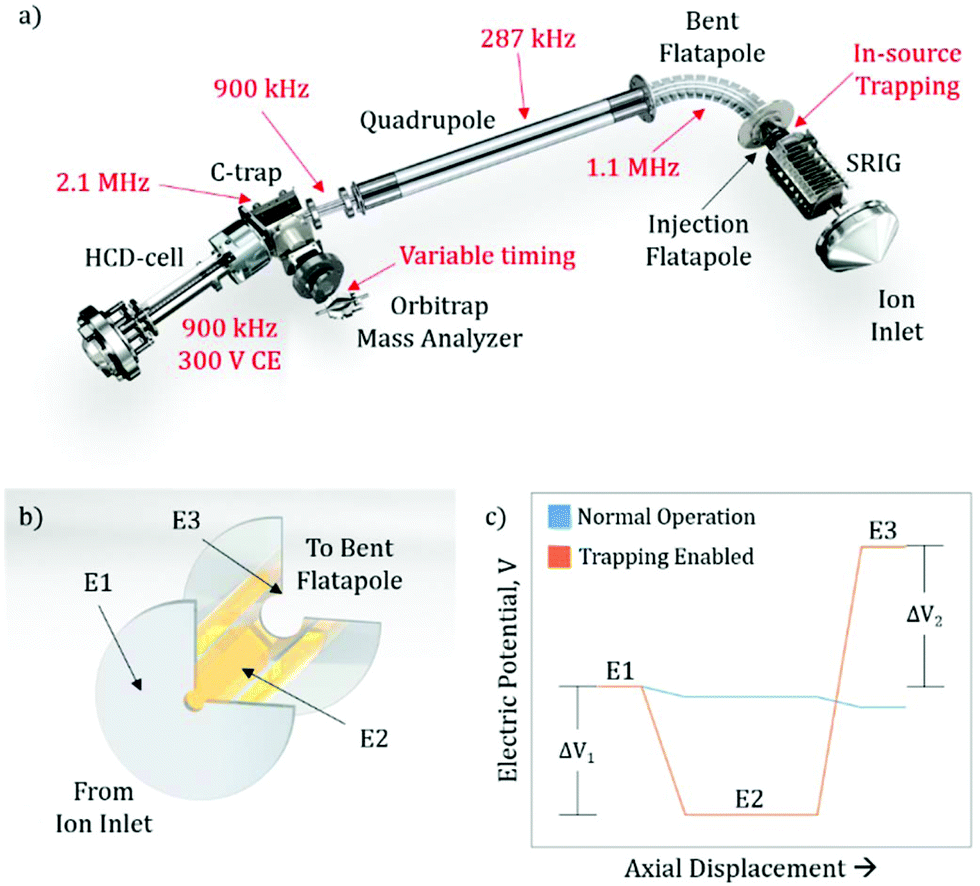 | ||
| Fig. 1 A schematic of the implementation of in-source trapping on the Q-Exactive Orbitrap mass spectrometer, (a), shows the various ion optic elements within the instrument and the modifications made in red. The schematic of the injection flatapole, (b), shows the SRIG exit lens (E1), injection flatapole (E2), and injection flatapole lens (E3). The relative electric potentials, (c), show that in the normal operation mode there is a slight decreasing gradient to guide the ions through this region. In the trapping enabled mode, these potentials are modified to allow for a desolvation voltage (ΔV1) and a trapping electric potential (ΔV2). | ||
Sample preparation and MS analysis
GroEL was prepared for native MS analysis by buffer exchanging the stock solution into 50 mM aqueous ammonium acetate at a pH of 6.8. After buffer exchange, the stock solution was diluted to a final concentration of ∼2 μM. Myoglobin (Sigma Aldrich, St Louis, MO) was prepared by diluting the sample to a final concentration of 2 μM. The capsid-forming enzyme lumazine synthase from the hyperthermophilic bacterium Aquifex aeolicus (AaLS) was mutated and purified as described previously (AaLS capsid protein (Cp); monomer mass = 16736.2 Da).15 The capsid samples were buffer exchanged into 150 mM ammonium acetate with 25 mM triethylammonium acetate, at a pH of 8.0 for charge reducing conditions and 150 mM ammonium acetate, pH 8.0 for non-charge reducing conditions. Aliquots of 1–2 μL of each analyte solution were loaded into a borosilicate capillary emitter that had been gold-coated. Ionization was facilitated by biasing this emitter +1.2–1.4 kV with respect to the heated capillary inlet. MS analysis was performed at a transient length of 64 ms for GroEL and AaLS. During comparative studies between in-source trapping enabled and normal operation, all MS instrument parameters, including RF ion optic frequencies, were held constant. The trapping potential was held at +69 V and the desolvation voltage was variable as noted in the text. For variable injection flatapole RF voltage studies, the RF Vp–p values were changed in the acquisition software. Mass resolution was calculated using the full width half maximum (FWHM) definition.
Results and discussion
Previous modifications to the Q Exactive Orbitrap mass spectrometer focused on lowering the RF frequency of the ion optics to allow for better transmission characteristics of high m/z ions.8,13 While further decreasing the frequency of other RF confining ion optics should also increase this m/z limit (see section: Pushing the detection limit of high m/z ions), it highlighted the limitations imposed by other elements within the instrument. One such restriction is the ion transmission through the bent flatapole region of the source (Fig. 1a), where the momentum the ion possesses from free-jet expansion during the transfer from atmospheric to high vacuum can propel the ion out of the stable transmission region and lead to an overall loss of sensitivity. To address this, we hypothesized that in-source ion trapping within the injection flatapole can be used to dampen the momentum of the ions prior to entering the bent flatapole region and increase the ion transmission efficiency. Moreover, through the use of variable voltage gradients, as the ions enter the in-source trapping region, improved desolvation can be implemented. Recently, it has been shown that modifications that utilized voltage gradients and in-source trapping can supply enough energy to fragment small protein complexes, allowing for pseudo-MS3 workflows; however, these modifications required extensive hardware modification to the source ion optics within the instrument.16 Here the modifications described are less invasive, i.e. no ion optics are being replaced, and should provide the desired improved desolvation, allow for pseudo-MS3 (see ESI:† MS3 capabilities on small and large protein assemblies), as well as, uniformity of ion transmission over a very wide m/z range due to the momentum dampening.Implementation of in-source trapping
To implement in-source trapping capabilities in the mass spectrometer, the electronics of the instrument were modified to allow for pulsing of the electrical potentials within the injection flatapole region of the source (Fig. 1a). With the modification, the applied voltage on the stacked ring ion guide (SRIG) exit lens (E1, Fig. 1b), injection flatapole (E2), and injection flatapole lens (E3) can be independently controlled and the source can be operated in two modes: normal operation and trapping enabled (Fig. 1c). In the normal operation mode, the voltage values are unmodified and a small, downhill potential gradient facilitates ion transmission through this region. In the trapping enabled mode, the DC bias of E2 is lowered so the ions are subjected to a negative potential gradient between E1 and E2, termed the desolvation voltage (ΔV1). Additionally, the DC potential of E3 is raised to form a potential barrier between E2 and E3, termed the trapping potential (ΔV2). This barrier blocks the ions from exiting the injection flatapole and thus enables their trapping within E2. When this occurs, ions undergo collisions with background gas, losing their initial momentum from free-jet expansion and get stored at the bottom of the potential well. After 4 ms, the electrical potentials are pulsed back to their normal operation values and the ions exit the injection flatapole. Both the desolvation voltage and the trapping potential are critical to improving the performance of the instrument, both in terms of mass resolution and ion transmission efficiency. The desolvation voltage is designed to remove non-specific solvent adducts, a primary limitation to obtainable resolution, while the trapping potential improves overall ion transmission and its uniformity in m/z by reducing the ions’ axial momentum to values around thermal ones.Improving ion transmission and mass resolution with in-source trapping
To characterize the increase in mass resolution and ion transmission that is obtained through the use of in-source trapping, GroEL (14-mer, theoretical MW = 800766.4 Da) was used as model system and analyzed in the two various modes of operation. Both of these spectra (Fig. 2a and b) were collected without use of the HCD cell to isolate the desolvation effect of the in-source trapping modification. The mass spectrum in normal operation mode shows a charge state envelope of the intact protein complex with charge states ranging from 71 to 64+ (Fig. 2a). Trapping enabled mode shows a similar charge state envelope (Fig. 2b); however, here the mass resolution of each charge state is substantially increased. For the 68+ charge state in normal operation, there are partially solvated ions, as seen by the presence of shoulder peaks that occur at 11820 m/z, which lead to considerable peak broadening (Fig. 2a, inset). Upon application of the desolvation voltage, the intensity of solvated ions is reduced, the peak width narrows, and the centroid is shifted to lower m/z values, consistent with more efficient desolvation. While desolvation is not complete, as shown by a peak width that is higher than the isotopically limited peak width (ca. 1 m/z), these data show that in-source trapping assists in desolvation, leading to an increase in the apparent mass resolving power from 1070 in normal operation mode to 1680 during trapping enabled mode (Fig. 2a and b, insets).
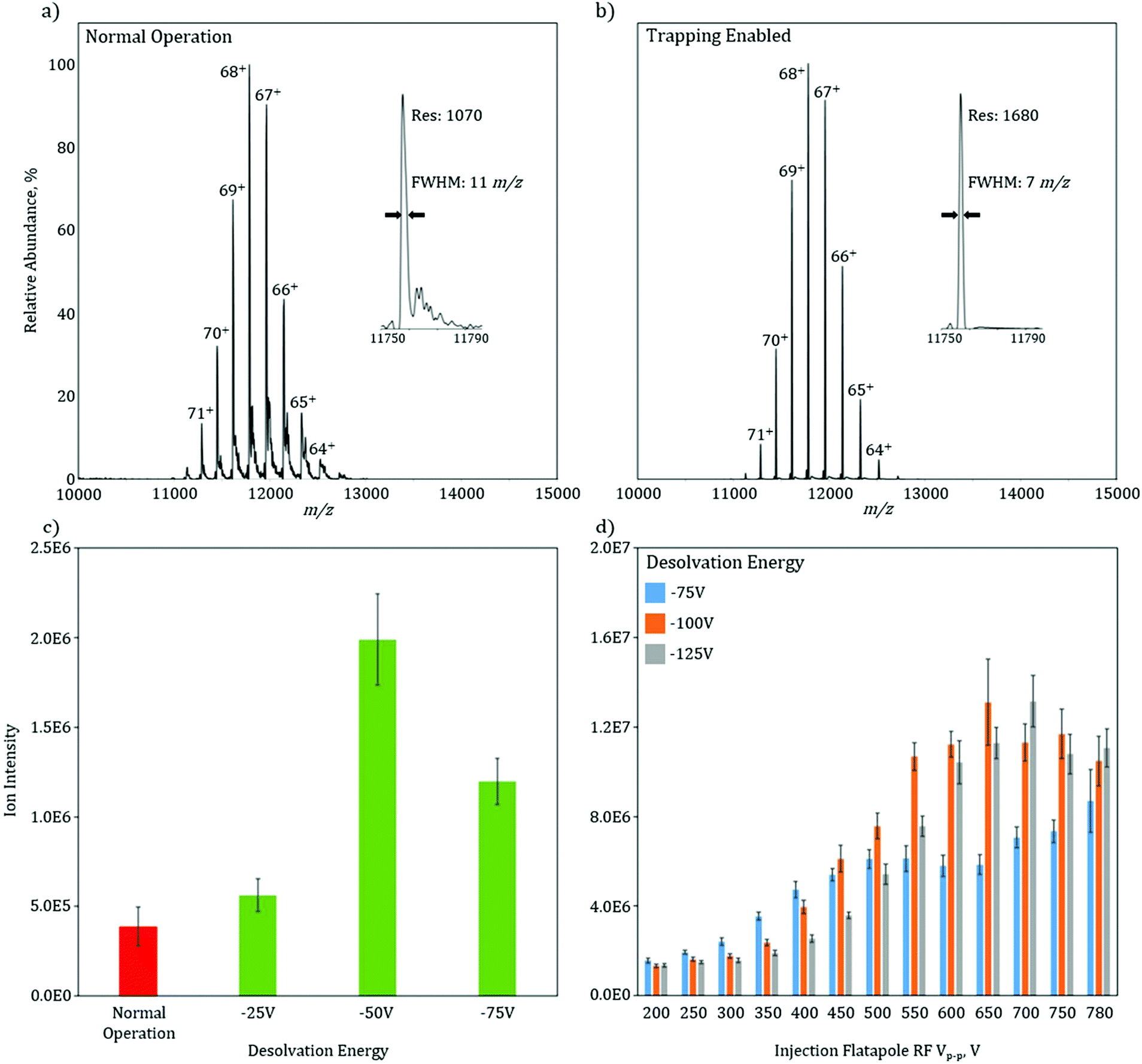 | ||
| Fig. 2 Native nESI mass spectra of GroEl collected at standard operation (a) and with trapping enabled (b). The mass spectra show similar charge state envelopes of the GroEl 14-mer; however, inspection of the peak width of the 68+ charge state (insets) indicates that the trapping enabled mode provides higher resolution relative to the standard operation mode owing to more efficient desolvation. The base peak intensity of GroEl (c) as a function of desolvation energy shows a fivefold signal increase upon inclusion of in-source trapping. The dependence of the total ion current (TIC) on desolvation energy and injection flatapole RF Vp–p (d) shows that with tuning of confining electric potentials, a 6–7 fold increase in signal is seen. The error bars displayed in (c) and (d) represent the standard deviation of technical replicate MS scans. | ||
The combination of the desolvation voltage and the trapping potential allows for significant increases in ion intensity owing to improvements in transmission efficiency (see ESI Fig. 3†). The signal intensity of the base peak for GroEL without in-source trapping has an intensity of 4E5 (Fig. 2c). With the trapping enabled and using a desolvation voltage setting of −25 V, this intensity is increased by 20%. Increasing the desolvation voltage to −50 V, shows a fivefold signal increase. However, when this voltage is increased further to −75 V, there is a reduction in intensity as compared to a setting of −50 V. One plausible cause for this is the ions are increasingly radially scattered by the saddle-shaped reflecting field when the desolvation energy is increased, as described in the ESI† of paper.13 If the RF potential is no longer sufficient to correct this radial scattering, the ion transmission will be reduced. To test this hypothesis, the total ion current (TIC) of GroEL was monitored as a function of applied RF voltage amplitude and desolvation energy (Fig. 2d). At the lowest RF voltage amplitude, 200 Vp–p, the TIC is independent of the desolvation voltage. As the RF voltage is increased to 350 Vp–p, the TIC for −75 V increases while the increases for −100 V and −125 V are minimal. This data indicate that the RF voltage is now at a level that can radially confine the ions after they are subjected to the −75 V desolvation voltage, but at higher values and thus higher degrees of radial scatter, the ions are not efficiently radially trapped. The data clearly shows that there is an optimal RF voltage for each desolvation energy (and also each m/z), consistent with the variable scattering energies associated with each individual setting. For −75 V, the signal is optimized and plateaus at an RF voltage of 500 Vp–p, while 550 Vp–p and 600 Vp–p are needed for desolvation energies of −100 V and −125 V, respectively. Taken together, these data indicate that the combination of more effective desolvation, as well as, the reduction of the ions’ axial momentum through in-source trapping are responsible for the substantial increases in signal.
These gains in signal intensity become progressively important as the size of the biological system under investigation increases and as the chemical and or structural nature of the system becomes increasingly more difficult, e.g., membrane proteins, protein-nucleic acid complexes and endogenous protein complexes where ion signal can be divided amongst several different proteoforms, limiting the ion intensity for any one specific proteoform. The challenge arises from the ion signal intensity often dictating transient length, and thus the mass resolution, at which a mass spectrum can be acquired. For many analytes, these high transient acquisitions are often prohibitively long and result in poor signal-to-noise. Thus, the analytical benefit for in-source trapping is two-fold. The modification assists in desolvation of the ions, increasing the experimental obtained resolution or GroEL by 60% as compared to the unmodified instrument, even without further desolvation in the HCD cell. Moreover, the increase in signal intensity obtained with in-source trapping provides a means for faster acquisitions at higher mass resolution settings, which will allow for greater elucidation of structural detail.
Pushing the detection limit of high m/z ions: detecting virus-like particle dissociation products up to m/z 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) 000
000
With previous modifications, the Orbitrap-based mass spectrometer could detect ion signal in MS1 up to about 20000 m/z and 35000 m/z on the MS2 level; however, low transmission of these ions prevented use of long transient lengths and limited sensitivity.13 Now, with in-source trapping and all the RF frequencies being reduced, the transmission of very high m/z ions to the C-trap has greatly increased. However, it is important to consider the requirements needed to successfully transmit high m/z ions from the C-trap into the mass analyzer. When any ion is ejected from the C-trap toward the mass analyzer, a deflector electrode turns the ions into the entrance slot of the Orbitrap. After a set amount of time, the deflector electrode is pulsed to a compensation voltage to minimize field perturbations within the device, allowing for successful mass analysis, but, in doing so, prevents any additional ions from entering. While the standard pulse time works well for the peptides, proteins, and small protein complexes, larger m/z ions, where their time of flight from the C-trap to the Orbitrap is longer, do not have time to enter the Orbitrap before the deflector is pulsed to the compensation voltage and the Orbitrap is closed. Thus, this pulse time must be lengthened to allow these high m/z ions to enter the Orbitrap. As such, the final modification was to allow for variable pulse times of the deflector electrode, keeping the Orbitrap “open” for longer and allowing the high m/z ions to enter. To investigate the cumulative effect of all the aforementioned modifications on the transmission properties of high m/z ions, mass analysis of a virus-like protein assembly was performed.
On the MS1 level, a homogeneous Cp 180-mer virus-like protein assembly under charge reducing conditions shows a well resolved charge state envelope centered around 30000 m/z (Fig. 3a), corresponding closely to the data previously reported by Sasaki et al. on the same system.15 To determine the maximum MS2m/z value achievable, the Cp 180-mer was analyzed with non-charge reducing conditions, which shifted the charge state envelope to 23000 m/z (Fig. 3b). Application of 250 V collision energy in the HCD-cell shows a product spectrum (Fig. 3c) with ion signals up to 50000 m/z, corresponding to the sequential loss of individual protein subunits (see Table 1 for mass assignments and resolution). At maximum collision energy (Fig. 3d), further subunit ejection produces well resolved ion signals up to 70000 m/z. In fact, at the highest m/z signal obtained (Fig. 3d, right inset), the mass resolution is still over 500, which allows for baseline separation of the individual dissociation products at these high m/z values (Fig. 3d, left inset). Furthermore, this increase in detectable m/z value almost doubles the previous limit of unmodified instrument and reduces the spectra acquisition time from what was previously hours, to minutes on the modified machine owing to higher transmission efficiency.
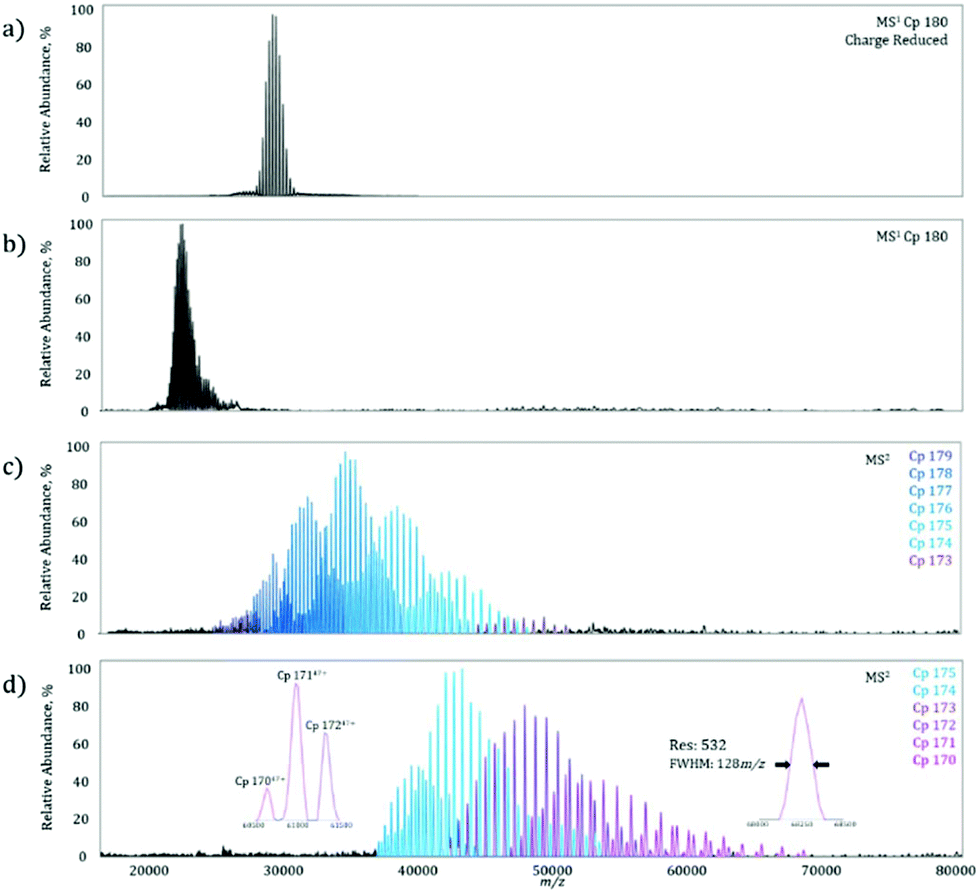 | ||
| Fig. 3 Analysis of MDa virus-like particles on the modified mass spectrometer. During an MS1 scan, the charge-reduced Cp180 (a) shows a well resolved charge state envelope centered on ∼30000 m/z. To further shift ions to higher m/z values and demonstrate the full capability of the instrument, the non-charge reduced Cp180 (b) charge state envelope was subjected to increasing HCD collision energies. At a collision energy of 250 V, the MS2 products extend to 50000 m/z (c). At maximal HCD, 300 V (d), the production mass spectrum shows further fragmentation of the Cp180 assembly and ions up 70000 m/z. The mass resolution is high enough to baseline-resolve these different dissociation products even at this high m/z value (d, left inset). At the highest m/z, the mass resolution is greater than 500 (d, right inset). | ||
| Ejected subunits | Deconvoluted mass (kDa) | Expected mass (kDa) | Deviation from expected mass | Average mass resolution | |
|---|---|---|---|---|---|
| Cp 180 | 0 | 3021.1 ± 0.4 | 3012.5 | 0.28% | 375 |
| Cp 179 | 1 | 3003.3 ± 0.4 | 2995.8 | 0.25% | 669 |
| Cp 178 | 2 | 2986.0 ± 0.3 | 2979.0 | 0.23% | 517 |
| Cp 177 | 3 | 2968.9 ± 0.2 | 2962.3 | 0.22% | 471 |
| Cp 176 | 4 | 2951.8 ± 0.2 | 2945.6 | 0.21% | 450 |
| Cp 175 | 5 | 2932.7 ± 0.3 | 2928.8 | 0.13% | 501 |
| Cp 174 | 6 | 2915.7 ± 0.3 | 2912.1 | 0.13% | 468 |
| Cp 173 | 7 | 2899.0 ± 0.1 | 2895.4 | 0.13% | 429 |
| Cp 172 | 8 | 2882.3 ± 0.1 | 2878.6 | 0.13% | 401 |
| Cp 171 | 9 | 2865.9 ± 0.4 | 2861.9 | 0.14% | 455 |
Conclusions
Here, we describe the new modifications to the Q-Exactive Plus Orbitrap mass spectrometer which allow for in-source trapping, reduced RF frequencies on several ion optic elements, and variable Orbitrap fill times. One of the major limitations to the analysis of biological systems with native MS has been the low transmission efficiency at high m/z, which limits obtainable sensitivity and resolution. With these modifications, signal transmission shows more than a fivefold increase and mass resolution increases by 60% for GroEL. Furthermore, the collective result of the modifications is an increase in high m/z limit of the instrument, with well resolved charge states up to 70000 m/z being shown. These attributes will become increasingly advantageous for the future analysis of certain biological systems, namely systems that possess a high degree of structural complexity. Many biological systems require binding of small molecule co-factors such as magnesium or adenosine triphosphate (ATP); moreover, endogenous protein complexes often exhibit can exhibit post translational modifications or site amino-acid point-mutations. Currently, these present a challenge for modern mass spectrometry as low signal intensity often requires the use of suboptimal mass resolution settings, limiting the depth of structural detail measured with native MS. Thus, the modifications described here provide the next stepping stone in instrumentation and open the door for faster, better-resolved native MS analysis of these systems.
Conflicts of interest
Kyle L. Fort, Dmitriy Boll, Maria Reinhardt-Szyba, Mikhail E. Belov, and Alexander A. Makarov are all employees of Thermo Fisher Scientific, the manufacturer and distributor of the Q Exactive Plus mass spectrometer.Acknowledgements
We acknowledge the support from the Netherlands Organization for Scientific Research (NWO) funding the large-scale proteomics facility Proteins@Work (project 184.032.201) embedded in the Netherlands Proteomics Centre. M. W. and A. J. R. H. are additionally supported by a Projectruimte grant (12PR3303-2) from Fundamenteel Onderzoek der Materie (FOM). K. F., A. M. and A. J. R. H. acknowledge additional support through the European Union Horizon 2020 programme FET-OPEN project MSmed, Project 686547. A. J. R. H. acknowledge further support by the NWO TOP-Punt Grant 718.015.003.Notes and references
- M. Wilm and M. Mann, Anal. Chem., 1996, 68, 1–8 CrossRef CAS PubMed.
- M. S. Wilm and M. Mann, Int. J. Mass Spectrom. Ion Processes, 1994, 136, 167–180 CrossRef CAS.
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CAS.
- H. Hernandez and C. V. Robinson, Nat. Protoc., 2007, 2, 715–726 CrossRef CAS PubMed.
- B. T. Ruotolo, J. L. P. Benesch, A. M. Sandercock, S.-J. Hyung and C. V. Robinson, Nat. Protocols, 2008, 3, 1139–1152 CAS.
- V. Katta and B. T. Chait, J. Am. Chem. Soc., 1991, 113, 8534–8535 CrossRef CAS.
- J. Snijder, R. J. Rose, D. Veesler, J. E. Johnson and A. J. R. Heck, Angew. Chem., Int. Ed., 2013, 52, 4020–4023 CrossRef CAS PubMed.
- R. J. Rose, E. Damoc, E. Denisov, A. Makarov and A. J. R. Heck, Nat. Methods, 2012, 9, 1084–1086 CrossRef CAS PubMed.
- C. Uetrecht, I. M. Barbu, G. K. Shoemaker, E. van Duijn and A. J. R. Heck, Nat. Chem., 2011, 3, 126–132 CrossRef CAS PubMed.
- J. Snijder and A. J. R. Heck, Annu. Rev. Anal. Chem., 2014, 7, 43–64 CrossRef CAS PubMed.
- A. J. R. Heck, Nat. Methods, 2008, 5, 927–933 CrossRef CAS PubMed.
- J. A. Loo, Mass Spectrom. Rev., 1997, 16, 1–23 CrossRef CAS PubMed.
- J. Snijder, M. van de Waterbeemd, E. Damoc, E. Denisov, D. Grinfeld, A. Bennett, M. Agbandje-McKenna, A. Makarov and A. J. R. Heck, J. Am. Chem. Soc., 2014, 136, 7295–7299 CrossRef CAS PubMed.
- M. van de Waterbeemd, K. L. Fort, D. Boll, M. Reinhardt-Szyba, A. Routh, A. Makarov and A. J. R. Heck, Nat. Methods, 2017, 14, 283–286 CrossRef CAS PubMed.
- E. Sasaki, D. Bohringer, M. van de Waterbeemd, M. Leibundgut, R. Zschoche, A. J. R. Heck, N. Ban and D. Hilvert, Nat. Commun., 2017, 8, 10 CrossRef PubMed.
- G. Ben-Nissan, M. E. Belov, D. Morgenstern, Y. Levin, O. Dyrn, G. Arkind, G. Lipson, A. A. Makarov and M. Sharon, Anal. Chem., 2017, 89, 4708–4715 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an01629h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |