
Polyacrylamide gel electrophoresis of semiconductor quantum dots and their bioconjugates: materials characterization and physical insights from spectrofluorimetric detection†
 a
and
W. Russ
Algar
*a
a
and
W. Russ
Algar
*a
Abstract
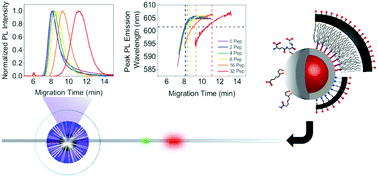
Colloidal semiconductor quantum dot (QD) nanocrystals have ideal fluorescence properties for bioanalysis and bioimaging, but these materials must be functionalized with an inorganic shell, organic ligand or polymer coating, and conjugated with biomolecules to be useful in such applications. Several different analytical techniques are used to characterize QDs and their multiple layers of functionalization. Here, we revisit poly(acrylamide) gel electrophoresis (PAGE), which has been scarcely used for the characterization of QDs and their bioconjugates in deference to the routine use of agarose gel electrophoresis. We implemented PAGE in a novel “stubby” capillary format with spectrofluorimetric detection, the combination of which enabled more rapid and more detailed characterization of QDs than was possible with both poly(acrylamide) and agarose slab gels. Correlations between the peak photoluminescence (PL) emission wavelength and electropherogram peaks, especially when combined with Ferguson analysis, provided new and significant insight into the key factors that determine the electrophoretic mobility of QDs, and helped to resolve heterogeneity and sub-populations in ensembles of QDs. The method was useful for characterization of the inorganic core/shell nanocrystals, their organic ligand and polymer coatings, and their final bioconjugates, the latter of which were in the form of peptide and protein conjugates. With further development and optimization, we anticipate that capillary PAGE with spectrofluorimetric detection will become a valuable addition to the toolbox of characterization techniques suitable for QDs, their bioconjugates, and other nanoparticle materials as well.
- This article is part of the themed collection: RSC papers by GRC Bioanalytical Sensors 2018 Speakers
 Please wait while we load your content...
Please wait while we load your content...

