
Distinction between PTB7-Th samples prepared from Pd(PPh3)4 and Pd2(dba)3/P(o-tol)3 catalysed stille coupling polymerization and the resultant photovoltaic performance†
Jianhong
Gao
a,
Wei
Wang
a,
Shoujie
Zhang
a,
Shengqiang
Xiao
 *a,
Chun
Zhan
a,
Mingyan
Yang
a,
Xinhui
Lu
*b and
Wei
You
*ac
*a,
Chun
Zhan
a,
Mingyan
Yang
a,
Xinhui
Lu
*b and
Wei
You
*ac
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, P. R. China. E-mail: shengqiang@whut.edu.cn
bDepartment of Physics, Chinese University of Hong Kong, Hong Kong, P. R. China. E-mail: xhlu@phy.cuhk.edu.hk
cDepartment of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, USA. E-mail: wyou@unc.edu
First published on 27th November 2017
Abstract
State-of-the-art polymer donors for bulk heterojunction (BHJ) polymer solar cells (PSCs) are mostly alternating donor–acceptor (D–A) copolymers prepared from palladium catalysed Stille cross-coupling condensations. The structural variation of D–A copolymers, such as conjugated backbones, alkyl side chains and positions, substituents and molecular weights, has been proven to significantly impact the energy levels, intermolecular interactions and molecular packing, which hereafter synergistically determine the performance of corresponding BHJ PSCs. For a given D–A copolymer, the alternation of D and A units does not always proceed as intended when a specific catalyst is employed. The actual D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A ratios and the molecular weights would most likely be inconsistent as well when catalyzed differently in preparation. To clarify the impact of the catalysts employed for polymerizations on the structure of the resultant polymers and on the corresponding photovoltaic performance, a comprehensive investigation was conducted on the distinction between two PTB7-Th samples prepared from Stille coupling polymerization with the classic palladium catalysts Pd2(dba)3/P(o-tol)3 and Pd(PPh3)4, respectively. The structural variation between the two PTB7-Th samples is discovered to be distinct with respect to both the actual D:A ratios and the molecular weights, which endow the two samples with entirely different aggregation behaviors and optoelectronic properties. The optimized polymer:PC71BM BHJ PSC device demonstrates a normal PCE of 8.65% with the PTB7-Th sample catalyzed by Pd(PPh3)4 as reported and a deteriorated PCE of 4.07% with the PTB7-Th sample catalyzed by Pd2(dba)3/P(o-tol)3. A significant morphological evolution between the two PTB7-Th samples from the neat film to the BHJ film was clarified. This wealth of information on the strong correlation among the variations in the chemical structure, the morphology and the device performance allows the establishment of guidance on the selection of the appropriate catalyst to obtain high-performance PSC polymers.
:A ratios and the molecular weights would most likely be inconsistent as well when catalyzed differently in preparation. To clarify the impact of the catalysts employed for polymerizations on the structure of the resultant polymers and on the corresponding photovoltaic performance, a comprehensive investigation was conducted on the distinction between two PTB7-Th samples prepared from Stille coupling polymerization with the classic palladium catalysts Pd2(dba)3/P(o-tol)3 and Pd(PPh3)4, respectively. The structural variation between the two PTB7-Th samples is discovered to be distinct with respect to both the actual D:A ratios and the molecular weights, which endow the two samples with entirely different aggregation behaviors and optoelectronic properties. The optimized polymer:PC71BM BHJ PSC device demonstrates a normal PCE of 8.65% with the PTB7-Th sample catalyzed by Pd(PPh3)4 as reported and a deteriorated PCE of 4.07% with the PTB7-Th sample catalyzed by Pd2(dba)3/P(o-tol)3. A significant morphological evolution between the two PTB7-Th samples from the neat film to the BHJ film was clarified. This wealth of information on the strong correlation among the variations in the chemical structure, the morphology and the device performance allows the establishment of guidance on the selection of the appropriate catalyst to obtain high-performance PSC polymers.
1. Introduction
Polymer solar cells (PSCs) are attracting an increasing amount of interest due to their potential for large-area and flexible applications via solution processing at low cost.1–8 The most effective and therefore extensively investigated device concept of PSCs is established on the bulk-heterojunction (BHJ) approach, in which a polymeric electron donor and an electron acceptor are blended to offer a single photoactive layer.9 The past few decades have witnessed tremendous effort in the design and synthesis of novel electron acceptors and polymer donors with optimized energy levels and band gaps for high power conversion efficiency (PCE). Fullerene derivatives such as PCBM and PC71BM represented some of the most successful electron acceptors before the emergence of efficient non-fullerene small molecule acceptors.10–12 Despite lagging behind their fullerene and small molecule counterparts in terms of performance, polymer acceptors have also shown rapid progress.13,14 Meanwhile, the development of highly efficient polymer donors has emerged as one of the main areas of focus in PSC research.15–20 The optimization of material design with synergetic efforts on device and interface engineering over the years has led to a record power conversion efficiency (PCE) of up to 12% for polymer:fullerene blends,21 13% for polymer:nonfullerene small molecule acceptor blends,22 and 9% for all-polymer blend BHJ PSCs in a single junction,23,24 respectively. These developments strongly enhance the potential of PSCs to become a real renewable energy conversion technology. On the other hand, it would be onerous as well to develop new polymer donors with further improved performance due to the predicament of predicting whether a given polymeric donor would offer high device performance.As acquired from a lot of BHJ PSC paradigms, each step within a photoelectric conversion process has been proven to be not only strongly correlated with the energy state of the materials employed (e.g., the energy level alignment of the pair of electron donor and acceptor) but also with the morphological structures (molecular packing and orientation and nanoscale phase separation) of the BHJ films.25 Constructing conjugated polymers with alternating electron-donating (D) and electron-accepting (A) units has proven to be the most powerful strategy for controlling energy due to their high flexibility in tuning the energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) and the band gaps of the resultant polymers.26 It therefore becomes essentially important to arrange molecules in a manner that optimizes all photoelectric conversion steps required for efficient device operation. However, the optimized arrangement of conjugated polymers in the solid state especially in a BHJ film is extremely sensitive to their chemical structures, surroundings and processing, making them unpredictable and can only be revealed by characterization after being processed.27 As such, tremendous efforts on molecular engineering of polymer donors have been invested to fundamentally characterize and understand the correlation between the chemical structure and the morphology in conjugation with the device characteristics.15–20,28 A number of performance-affecting and molecular structure related variables have been disclosed and intensively investigated, such as conjugated backbones,16,18–20 alkyl side chains and positions,29–31 substituents,32,33 molecular weight, polydispersity (PDI), etc.34–36 The conjugated backbone dominates the energy levels and the band gap. The alkyl side chains linked to the backbone largely determine the solubility in the processing solvent. The substituents linked to the backbone with varied electron accepting/donating ability and steric interactions assist in adjusting the energy levels. Most importantly, all of these structural variables also have a significant impact on the intermolecular interactions, molecular packing and phase separation in the BHJ blend and they tend to function synergistically in a PSC device.
State-of-the-art polymer donors are mostly D–A copolymers typically prepared from palladium catalysed Stille cross-coupling condensations between an aryl halide (e.g., Br or I) and an organotin monomer. Ideally, this synthetic approach yields perfectly alternating copolymers because the sp2 carbon–halide bond can only react with an organotin bond (sp2 C–Sn bond) and vice versa.37 However, the alternation of D and A units does not always proceed as intended due to the existence of homocoupling side reactions when a specific palladium catalyst is employed,38,39 let alone in the case of different catalysts. Moreover, the molecular weights and PDIs of the polymers with the same alternating D and A units could most likely be inconsistent as well with the utilization of different catalysts in preparation. It needs to be noted that so far most of the D–A copolymers with varied molecular weights for PSCs have been obtained mainly by controlling either the reaction time or the feed ratio of D and A units under the same reaction conditions. These structural variations of D–A copolymers caused by the use of different catalysts for the same polymerization reaction will obviously impact the corresponding PSC device performance.40 However, a systematic comparison of such impact on device performance has seldom been reported.
Due to the superior performance in polymer:fullerene BHJ PSCs with a stable PCE in the range of 8–10%,17 PTB7-Th has been extensively and successfully employed as a perfect donor to identify prominent acceptors. The reported preparation of PTB7-Th was achieved via Pd(PPh3)4 catalysed Stille coupling polymerization between the donor monomer of (4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl)bis(trimethylstannane) (BDTT) and the acceptor monomer of 2-ethylhexyl-4,6-dibromo-3-fluorothieno[3,4-b]thiophene-2-carboxylate (FTT).41 It is also discovered that most of the high-performance D–A copolymers (with PCE > 10% for the corresponding PSCs) were prepared by using palladium catalysts of either Pd2(dba)3/P(o-tol)3 or Pd(PPh3)4, for example, PBnDT-FTAZ,42,43 PNTT-H,44 PvBDTTAZ,45 the PffBT-4T series, etc. catalysed by Pd2(dba)3/P(o-tol)3,31 while J71,46 PNTz4TF2,47 PBDBT-SF, etc. catalysed by Pd(PPh3)4.22 To clarify the impact of the two classic catalysts employed for the polymerizations on the structure of the resultant polymers and corresponding photovoltaic performance, in this contribution, we present a comprehensive investigation on the distinction between the PTB7-Th samples prepared from Stille coupling polymerization catalysed by Pd2(dba)3/P(o-tol)3 and Pd(PPh3)4, respectively. The two samples of PTB7-Th were accordingly coded PTB7-Th-L and PTB-Th-H in this work. Indeed, the variation of chemical structures between the two PTB7-Th samples is disclosed to be distinct with respect to both the actual ratios between D and A units and the molecular weights as characterized by 1H NMR, Raman spectra and high temperature gel permeation chromatography (GPC) techniques. The standard polymer:PC71BM blend BHJ PSC fabricated with PTB7-Th-H demonstrates a high and normal PCE of 8.65% as reported. Nevertheless, utilizing the catalyst of Pd2(dba)3/P(o-tol)3 leads to the sample of PTB7-Th-L with a seriously deteriorated PCE of 4.07%. To gain an in-depth understanding of the gap of photovoltaic performance, the photo-induced charge generation, charge transport and recombination within the devices are investigated in combination with the morphological characterization of the two PTB7-Th samples both in neat and BHJ blend films. A strong correlation among the variations in the chemical structure, the morphology and the device performance is observed.
2. Experimental section
2.1 Morphological characterization
AFM morphology measurement of the films was carried out in tapping mode on a Multimode 8 SPM under ambient conditions. RTESPA (0.01–0.025 ohm cm antimony (n) doped silicon) tips with a spring constant of 20–80 N m−1 and a frequency of 305–356 kHz were used in imaging. The grazing incidence wide angle X-ray scattering (GIWAXS) measurement of the pristine polymers and the BHJ blend films was conducted at BL23A1 of the National Synchrotron Radiation Research Center, Hsinchu, Taiwan.2.2 Device fabrication and characterization
The PSC devices were fabricated with the inverted structure of ITO/ZnO (∼35 nm)/polymer:PC71BM/MoO3(∼8 nm)/Ag(100 nm). The ZnO sol–gel solution was spin-coated at 4000 rpm for 40 seconds on a pre-cleaned ITO glass substrate. The film was then heated at 180 °C for 40 min in air to obtain the electron transport layer of ZnO with an ∼35 nm thickness before being transferred to a glovebox. The polymer:PC71BM (1:1.5, w/w) blend film was spin-cast on the ZnO layer from the solution with a fixed donor concentration of 10 mg mL−1 in o-dichlorobenzene/1,8-diodooctane (97:3, v/v) and dried naturally in the glovebox. Finally, MoO3 (∼8 nm) and Ag (100 nm) were sequentially thermally evaporated and deposited on the top of active layers under a high vacuum of ∼2 × 10−6 mbar through a shadow mask defining 8 devices with each device area of 9 mm2. The thickness of the films was determined using a DEKTAK XT profilometer.
The current–voltage measurements were carried out in a glovebox under AM 1.5G irradiation (100 mW cm−2) from a 450 W solar simulator (Newport 94023A-U) calibrated using a NREL certified standard silicon cell. Current versus potential (J–V) curves were recorded with a Keithley 2420 digital source meter. External quantum efficiency (EQE) spectra were recorded as a function of wavelength from 300 to 900 nm on a Keithley 2400 source meter under the irradiation of a 300 W xenon lamp fitted with a 7-SCSpec spectral performance solar cell test system. The calibration of the incident monochromatic light was carried out with a Hamamatsu S1337-1010 BQ Silicon photodetector.
Electron and hole mobility of the blend film were tested by the space charge limited current (SCLC) method using ITO/ZnO (35 nm)/polymer:PC71BM/Ca (30 nm)/Al (80 nm) and ITO/PEDOT:PSS (40 nm)/polymer:PC71BM/MoO3 (8 nm)/Ag (100 nm) respectively by recording the dark current–voltage and applying sufficient voltage to form the space limited current.
3. Results and discussion
3.1 Structural characterization
The copolymers of PTB7-Th-L and PTB7-Th-H were synthesized via palladium catalysed Stille cross-coupling polycondensation reactions between BDTT and FTT monomers in a 1:1 feed ratio according to general reaction conditions and purification procedures for state-of-the-art PSC materials. Specifically, PTB7-Th-L was prepared by using chlorobenzene as the solvent and Pd2(dba)3/P(o-tol)3 as the catalyst while PTB7-Th-H was synthesized by using Pd(PPh3)4 as the catalyst in a mixed solvent of toluene:DMF (5:1, v/v) as reported.41 Soxhlet purification afforded the PTB7-Th-L sample from the CH2Cl2 fraction with an 85% yield. Nevertheless, the final product of PTB7-Th-H was obtained from the CHCl3 fraction with only a 58% yield. Both the PTB7-Th samples display high solubility in common organic solvents (e.g., CHCl3, toluene, chlorobenzene and o-dichlorobenzene) at room temperature. The thermal properties of PTB7-Th-L and PTB7-Th-H were investigated using thermogravimetric analysis (TGA) (Fig. S1†). Both samples exhibit sufficient thermal stability with loss of weight less than 5% on heating to 370 °C.
The alternating structure between BDTT and FTT units of both samples was confirmed by 1H NMR spectra (Fig. S2†). Due to distinctive chemical shifts of protons between α-CH2 linked to thiophenes on BDTT units (∼2.95 ppm, labeled a in Fig. S2†) and –OCH2 of the ester groups on FTT units (∼4.3 ppm, labeled b in Fig. S2†), we can easily figure out the actual D:A ratios between BDTT and FTT units within the conjugated backbones. It is found to be 1:0.97 for PTB7-Th-L and 1:1.1 for PTB7-Th-H, which deviated from the feed ratio of 1:1 for both copolymers. In other words, the proportion of the amount of FTT units in PTB7-Th-H is a little bit more than that of BDTT units while it is slightly smaller in PTB7-Th-L. The FTT unit has been regarded as a prequinoid aromatic unit able to effectively lower the band gap by dearomatizing to adopt a quinoid structure when incorporated into a conjugated polymer.26 Thus the incompatibility of the actual D:A ratios between the two PTB7-Th samples may lead to a change in optical properties.48 The number-average molecular weight (Mn) and the weight-average molecular weight (Mw) were evaluated by high temperature GPC using 1,2,4-trichlorobenzene as the eluent at 150 °C (Fig. S3†). The Mn and the Mw were determined to be 12.4 and 34.8 kDa for PTB7-Th-L with a PDI of 2.8 and 22.6 and 47.4 kDa for PTB7-Th-H with a smaller PDI of 2.1, respectively. Please note that the Mn of PTB7-Th-H is almost twice as high as that of PTB7-Th-L, indicative of a longer conjugation length.
For a specific conjugated polymer containing thiophene rings, the conjugated backbone vibrational modes such as symmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C stretching modes of the thiophene rings are Raman active due to their strong coupling with the π-electrons delocalized along the conjugated backbone. This feature makes them sensitive to the conjugation length and molecular planarity, which is valuable to gain insight into chemical structure variations. As depicted in Fig. 1, PTB7-Th-L exhibits a distinctive Raman spectrum with major peaks from vibrational modes of the conjugated backbone. Based on the referenced vibrational modes of P3HT and PTB7,49,50 peak 1 at 1462 cm−1 could be assigned to the CC stretching mode of the two free thiophenes in BDTT. The CC stretching mode of the fused thiophenes in BDTT could be regarded as peak 2 at 1494 cm−1. Peak 3 at 1529 cm−1 is a coupled vibration of the same mode to the CC stretching mode of the non-fluorinated thiophene in FTT, and peak 4 at 1572 cm−1 is the quadrant stretching mode coupled to the CC stretching mode of the fluorinated thiophene in FTT. A weak peak at ∼1721 cm−1 marked as peak 5 corresponds to the less Raman-active CO stretching mode of the ester chains in FTT. Although both polymers exhibit similar shapes in their CC and CO stretching vibrational modes, PTB7-Th-H shows a remarkable difference in the Raman vibrational modes in terms of a much increased intensity at peak 3 (∼1537 cm−1) and peak 4 (∼1578 cm−1) with vibrational modes shifted ∼8 and 6 cm−1 to higher wavenumbers, respectively. Moreover, peak 1 at 1467 cm−1 and peak 2 at 1499 cm−1 of PTB7-Th-H can also be observed with both vibrational modes shifted ∼5 cm−1 to higher wavenumbers and a slightly increased intensity. The CO stretching mode of the ester side chains in the FTT unit for PTB7-Th-H also exhibits a slightly increased intensity and a slight shift of ∼2 cm−1 to a lower wavenumber of 1719 cm−1. The much increased intensity and relatively larger shift to higher wavenumbers of peak 3 and 4 in PTB7-Th-H could be ascribed to the higher proportion of the amount of FTT loaded into the polymer backbone and longer conjugation length of the polymer as indicated above.
C and C–C stretching modes of the thiophene rings are Raman active due to their strong coupling with the π-electrons delocalized along the conjugated backbone. This feature makes them sensitive to the conjugation length and molecular planarity, which is valuable to gain insight into chemical structure variations. As depicted in Fig. 1, PTB7-Th-L exhibits a distinctive Raman spectrum with major peaks from vibrational modes of the conjugated backbone. Based on the referenced vibrational modes of P3HT and PTB7,49,50 peak 1 at 1462 cm−1 could be assigned to the CC stretching mode of the two free thiophenes in BDTT. The CC stretching mode of the fused thiophenes in BDTT could be regarded as peak 2 at 1494 cm−1. Peak 3 at 1529 cm−1 is a coupled vibration of the same mode to the CC stretching mode of the non-fluorinated thiophene in FTT, and peak 4 at 1572 cm−1 is the quadrant stretching mode coupled to the CC stretching mode of the fluorinated thiophene in FTT. A weak peak at ∼1721 cm−1 marked as peak 5 corresponds to the less Raman-active CO stretching mode of the ester chains in FTT. Although both polymers exhibit similar shapes in their CC and CO stretching vibrational modes, PTB7-Th-H shows a remarkable difference in the Raman vibrational modes in terms of a much increased intensity at peak 3 (∼1537 cm−1) and peak 4 (∼1578 cm−1) with vibrational modes shifted ∼8 and 6 cm−1 to higher wavenumbers, respectively. Moreover, peak 1 at 1467 cm−1 and peak 2 at 1499 cm−1 of PTB7-Th-H can also be observed with both vibrational modes shifted ∼5 cm−1 to higher wavenumbers and a slightly increased intensity. The CO stretching mode of the ester side chains in the FTT unit for PTB7-Th-H also exhibits a slightly increased intensity and a slight shift of ∼2 cm−1 to a lower wavenumber of 1719 cm−1. The much increased intensity and relatively larger shift to higher wavenumbers of peak 3 and 4 in PTB7-Th-H could be ascribed to the higher proportion of the amount of FTT loaded into the polymer backbone and longer conjugation length of the polymer as indicated above.
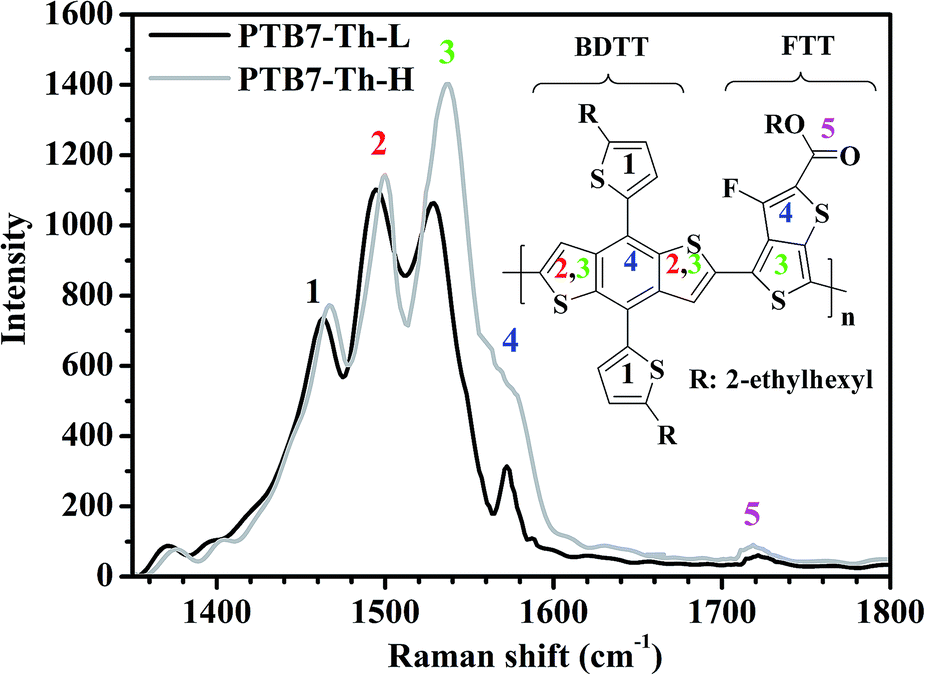 | ||
| Fig. 1 Raman resonance spectra of the PTB7-Th-L and PTB7-Th-H films excited at 514 nm under nitrogen (the inset is the chemical structure of PTB7-Th and colored numbers indicate the assignment of Raman modes to the vibrations of CC and CO bonds within the PTB7-Th backbone). | ||
3.2 Optical and electrochemical properties
The UV-vis absorption spectra of the two copolymers in diluted o-DCB solution with a concentration of 1 × 10−5 g mL−1 at varying temperatures and in film at room temperature are compiled in Fig. 2. As can be seen from Fig. 2a, PTB7-Th-L is well dissolved at 100 °C and a bathochromic shift of ∼20 nm can be observed for the maximum absorption wavelength (λmax) at ∼610 nm when the temperature decreases from 100 °C to room temperature. Moreover, the absorption spectrum of the PTB7-Th-L thin film shows a λmax of 648 nm with a redshift of ∼38 nm to that of the one in solution at 100 °C (Fig. 2d) and without the appearance of obvious fine structures. PTB7-Th-H is found to be well dissolved at elevated temperatures over 90 °C with λmax at ∼625 nm in the solution. In contrast, as the temperature decreases from 100 °C to room temperature, the presence of highly ordered preaggregates with strong π–π stacking is reflected in the redshifted absorption profile with the emergence of a strong and sharp transition at ∼700 nm, while the λmax of the conjugated backbone moves to ∼637 nm with a slight bathochromic shift of around 12 nm. Interestingly, the absorption spectrum of the PTB7-Th-H in film presents further a redshift of ∼10 nm to that in diluted solution at room temperature. Such strong preaggregation behaviour in diluted solution for PTB7-Th-H could possibly yield a highly ordered solid thin film. By monitoring the relative intensity of the transition peak as a function of temperature (normalized to the intensity observed at room temperature as shown in Fig. 2c), the order–disorder transition temperature is determined to be 90 °C. At an elevated temperature of 100 °C where PTB7-Th-H disaggregated, the absorption spectrum exhibits a slight red shift of ∼15 nm relative to that of diluted PTB7-Th-L solution at the same temperature, which could be attributed to the combined effect of higher molecular weight and higher FTT loading in the conjugated backbone.48 It is important to note that a similar λmax corresponding to the absorption of the conjugated backbone can be observed at 648 nm for both PTB7-Th samples in thin film. Moreover, PTB7-Th-H tends to give a slightly narrower spectrum with relatively intensified peaks in the absorption range both in solution and in film as shown in Fig. 2d, further suggesting an improved molecular ordering for PTB7-Th-H.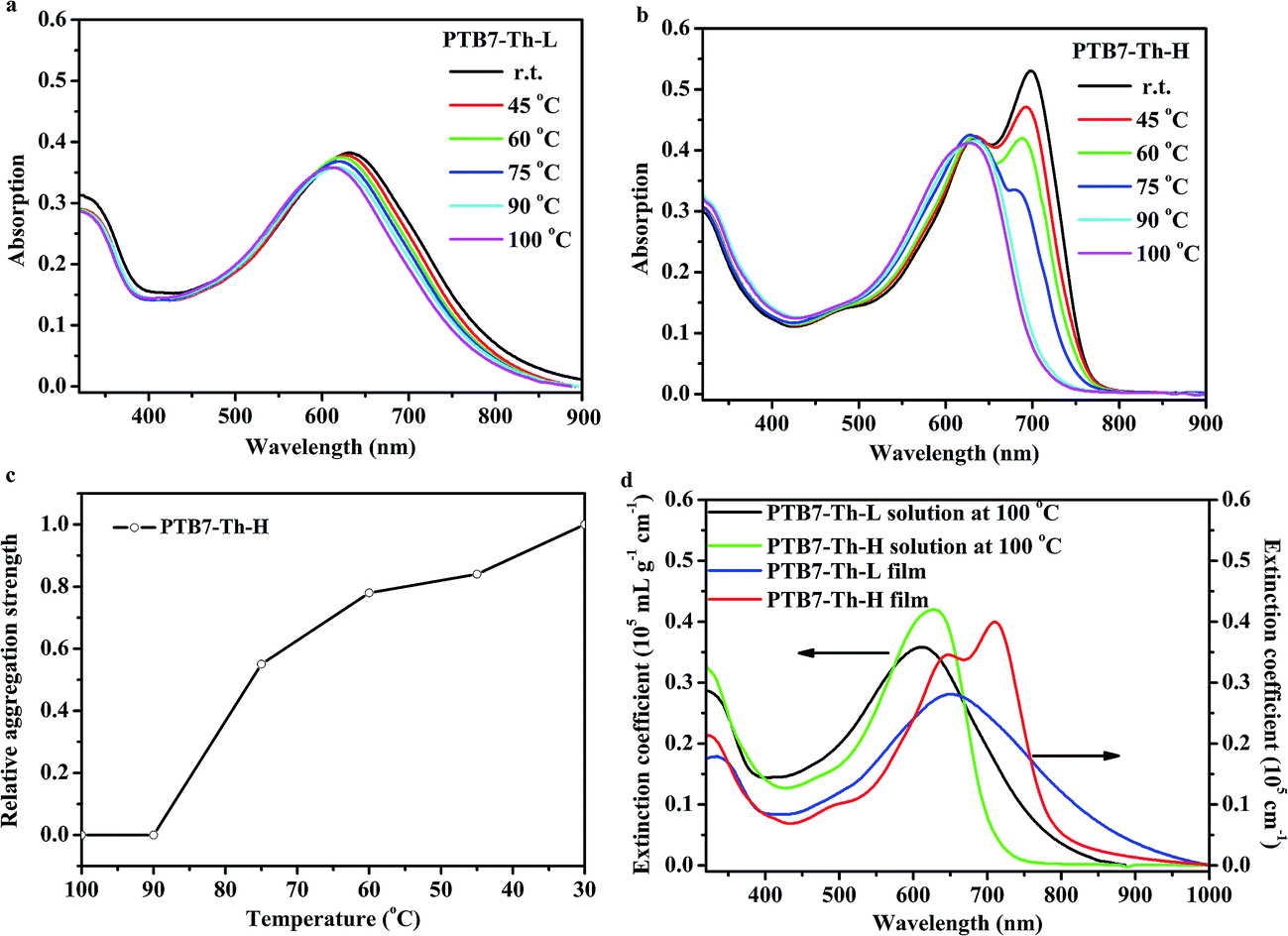 | ||
| Fig. 2 Temperature-dependent UV-vis absorption spectra of (a) PTB7-Th-L and (b) PTB7-Th-H solution in o-DCB (1 × 10−5 mg mL−1). (c) Plot of “relative aggregation strength” versus solution temperature. (d) Absorption coefficient spectra of PTB7-Th-L and PTB7-Th-H both in o-DCB solution (1 × 10−5 mg mL−1) at 100 °C and in film at room temperature. | ||
The optical band gaps (Eg) of PTB7-Th-L and PTB7-Th-H were calculated to be 1.43 eV from the absorption onset (λonset) at 870 nm and 1.59 eV from λonset at 780 nm in film, respectively. The highest occupied molecular orbital (HOMO) energy levels at −5.14 eV for PTB7-Th-L and −5.22 eV for PTB7-Th-H were calculated from the oxidation onset potentials from a cyclic voltammetry experiment as shown in Fig. S4.† The deeper-lying HOMO energy level of PTB7-Th-H can contribute to a higher open-circuit voltage (Voc) in PSCs when blending with a fullerene acceptor. Taking into account the optical band gap derived from the absorption onset in films, the LUMO energy levels of PTB7-Th-L and PTB7-Th-H were estimated to be −3.72 eV and −3.64 eV according to ELUMO = (EHOMO + Eg) eV respectively.
3.3 Solid state ordering of the pure polymers
Having observed significant variation in the absorption properties between PTB7-Th-L and PTB7-Th-H and their distinct preaggregation behaviour in diluted solution, we firstly tried powder X-ray diffraction (XRD) to gain insights into the structural order in the solid state. As seen from the XRD profiles of the two polymers in Fig. S5,† the PTB7-Th-L and PTB7-Th-H powders exhibit a reflection peak at a 2θ angle of around 3.82° and 3.95°, respectively, indicative of a lamellar value of 23.10 Å and 22.34 Å between polymer backbones separated by the flexible side chains ((100) diffraction). Both polymers show a diffraction peak at a 2θ angle of around 22.36° (a d-spacing of 3.97 Å), a typical π–π stacking spacing of conjugated backbones ((010) diffraction). Although the discrepancy of the lamellar and π–π stacking distances of both polymers is negligible, we can still notice slightly stronger diffractions from the PTB7-Th-H powder. As the aggregation and subsequent crystallization is presumed to be the main phase separation driving force in the blend film in a BHJ PSC device, we further employed GIWAXS to probe the molecular packing in the neat polymer films processed from 10 mg mL−1o-DCB solutions with 3% DIO under reported standard PSC device fabrication conditions for PTB7-Th. The GIWAXS results of neat PTB7-Th-L and PTB7-Th-H films are depicted in Fig. 3 (GIWAXS patterns are shown in Fig. S6†). Specifically, PTB7-Th-L and PTB7-Th-H present apparent (100) diffractions along the diffraction vector (q) both at ∼0.27 Å−1 in the in-plane (IP) direction, corresponding to a d-spacing value of ∼23.26 Å, and partially missing corresponding features in the out-of-plane (OOP) direction. Obvious (010) diffractions can only be observed in the OOP direction at the same q value of around 1.58 Å−1 (a d-spacing of 3.97 Å) for both films. However, the intensity of the (010) diffraction is a little bit stronger for the PTB7-Th-H film, indicative of a better crystallinity in accord with the result from the powder XRD testing. Briefly, the results indicate that the films of both polymers processed under reported PSC device fabrication conditions exhibit similar molecular ordering with particularly strong IP (100) and OOP (010) features, demonstrating a preferential face-on oriented π–π stacking of conjugated backbones.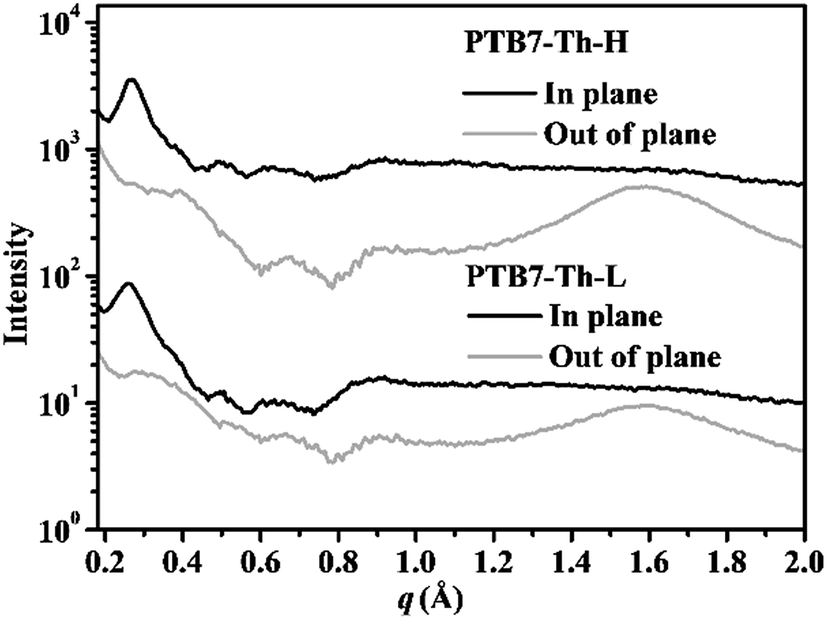 | ||
| Fig. 3 In-plane and out-of-plane line profiles of GIWAXS measurements on PTB7-Th-L and PTB7-Th-H films processed from 10 mg mL−1o-DCB solution with 3% DIO. | ||
3.4 Photovoltaic properties
PSC devices with the inverted structure of glass/ITO/ZnO/polymer:PC71BM (1:1.5 w/w)/MoO3/Ag were fabricated to enable direct comparison between the device performances of both PTB7-Th samples. The optimized weight ratio of polymer:PC71BM in the blends, the volume percentage of the processing additive of 1,8-diiodooctane (DIO), and the film thickness of the blends are found to be similar to those reported for PTB7-Th.41 The characteristic current density–voltage (J–V) curves of the optimized devices under simulated AM 1.5G illumination for both polymers are presented in Fig. 4a, and the corresponding photovoltaic parameters are detailed in Table 1. A maximum PCE of 8.65% was achieved using PTB7-Th-H with a Voc of 0.80 V, a short-circuit current density (Jsc) of 17.25 mA cm−2 and a fill factor (FF) of 63%, which normally lies in the efficiency range reported in the literature.17 The best PCE of 4.07% was obtained for the PTB7-Th-L based device with a Voc of 0.72 V, a Jsc of 10.81 mA cm−2 and an FF of 52%, considerably lower than that achieved for PTB7-Th-H. It is worth noting that batch-to-batch samples of the two polymers were also tried and analogous photovoltaic performances were obtained.
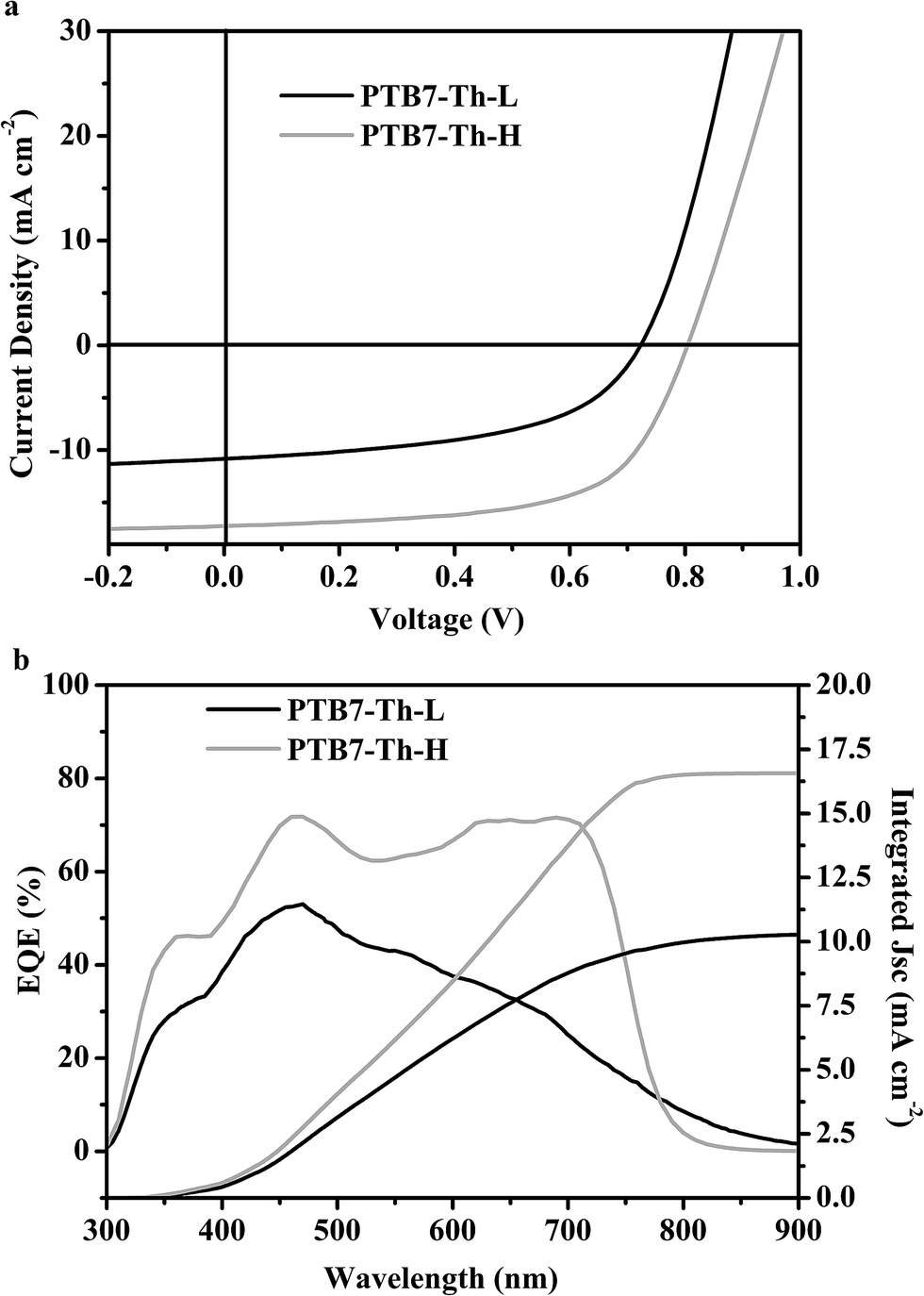 | ||
| Fig. 4 (a) Typical J–V curves of BHJ PSCs with the blend of PTB7-Th-L:PC71BM and PTB7-Th-H:PC71BM and (b) the corresponding EQE spectra and integrated Jsc curves. | ||
| Polymer | D:A weight ratio |
DIO (v/v) | Thickness (nm) | V oc (V) | J sc (mA cm−2) | FF | PCE (%) |
|---|---|---|---|---|---|---|---|
| a Calculated from EQE spectra as shown in Fig. 4b. b Values in parentheses are for average PCEs (over 10 devices). | |||||||
| PTB7-Th-L | 2:3 |
3% | 105 | 0.72 | 10.81(10.27)a | 0.52 | 4.07(3.85)b |
| PTB7-Th-H | 2:3 |
3% | 102 | 0.80 | 17.25(16.40)a | 0.63 | 8.65(8.50)b |
The external quantum efficiency (EQE) spectra of the BHJ devices for both polymers are depicted in Fig. 4b. The Jsc values calculated from the EQE spectra are consistent with those obtained from direct photo J–V measurements (Table 1). The significant changes of the Jsc values are well-reflected in their spectral responses from EQE curves. The PSC devices of both polymers yield broad EQE spectra from 300 to 800 nm. The EQE approaches 70% in the main absorption range from 450 to 720 nm for PTB7-Th-H based device while it is much lower for the device based on PTB7-Th-L, not surprising considering the fact that the PTB7-Th-H:PC71BM blend offered much improved light absorption as shown in Fig. S7.†
3.5 Charge generation, transport and recombination
It is recognized that the dramatic enhancement of the PCE value is attributed to the simultaneous increase of the Voc (∼10%), the FF (∼20%) and the Jsc (∼60%) for the PTB7-Th-H based device in comparison with PTB7-Th-L. The increase of the Voc can be reasonably ascribed to the deeper-lying HOMO energy level of PTB7-Th-H. The substantial increase of the Jsc and FF values for PTB7-Th-H suggests highly efficient charge generation, transport and collection steps within the PTB7-Th-H based device besides stronger light absorption of the blend film as mentioned above.51 In order to better understand the difference in photovoltaic performance between PTB7-Th-L and PTB7-Th-H, the charge generation, transport and charge recombination of the devices based on both polymers were then systematically investigated.We first measured the maximum photoinduced carrier generation rate (Gmax) in both devices under AM 1.5G illumination.52Fig. 5a reveals how the photogenerated current density (Jph) responded to the internal voltage (Vint) of the devices consisting of the two polymers. Jph increases in proportion to the voltage at low Vint, but saturates at high Vint (around 1.2 V and above, corresponding to a saturated photocurrent density defined as Jph,sat) where the internal field is large enough to sweep out all carriers to the electrodes. The Jph,sat value is determined to be 179.36 A m−2 for the device with PTB7-Th-H and 117.05 A m−2 for the device with PTB7-Th-L. The value of Gmax is calculated to be 0.72 × 1028 m−3 s−1 for the device with PTB7-Th-L, while the Gmax of the device with PTB7-Th-H is determined to be 1.07 × 1028 m−3 s−1 with a nearly 50% increase, according to Gmax = Jph,sat/(qL) where q is the elementary charge and L is the film thickness of the BHJ films. Obviously, the PTB7-Th-H based device offers more photogenerated excitons and dissociated charge carriers than the PTB7-Th-L based one, in great agreement with the result from fluorescence quenching experiments. As shown in Fig. S8,† the strong fluorescence of PTB7-Th-H with a maximum at ∼810 nm (excited at 688 nm) was dramatically quenched after mixing with PC71BM (2:3, w/w) into the BHJ film. A much higher quenching efficiency of up to 98% was obtained from the PTB7-Th-H:PC71BM blend compared to 86% from the PTB7-Th-L:PC71BM blend, indicating a much more efficient electron transfer process from PTB7-Th-H to PC71BM. In addition, a better diode quality in the dark J–V curve is achieved in the device consisting of PTB7-Th-H with a higher rectification factor (between −3 V and +3 V) and a lower leakage current.
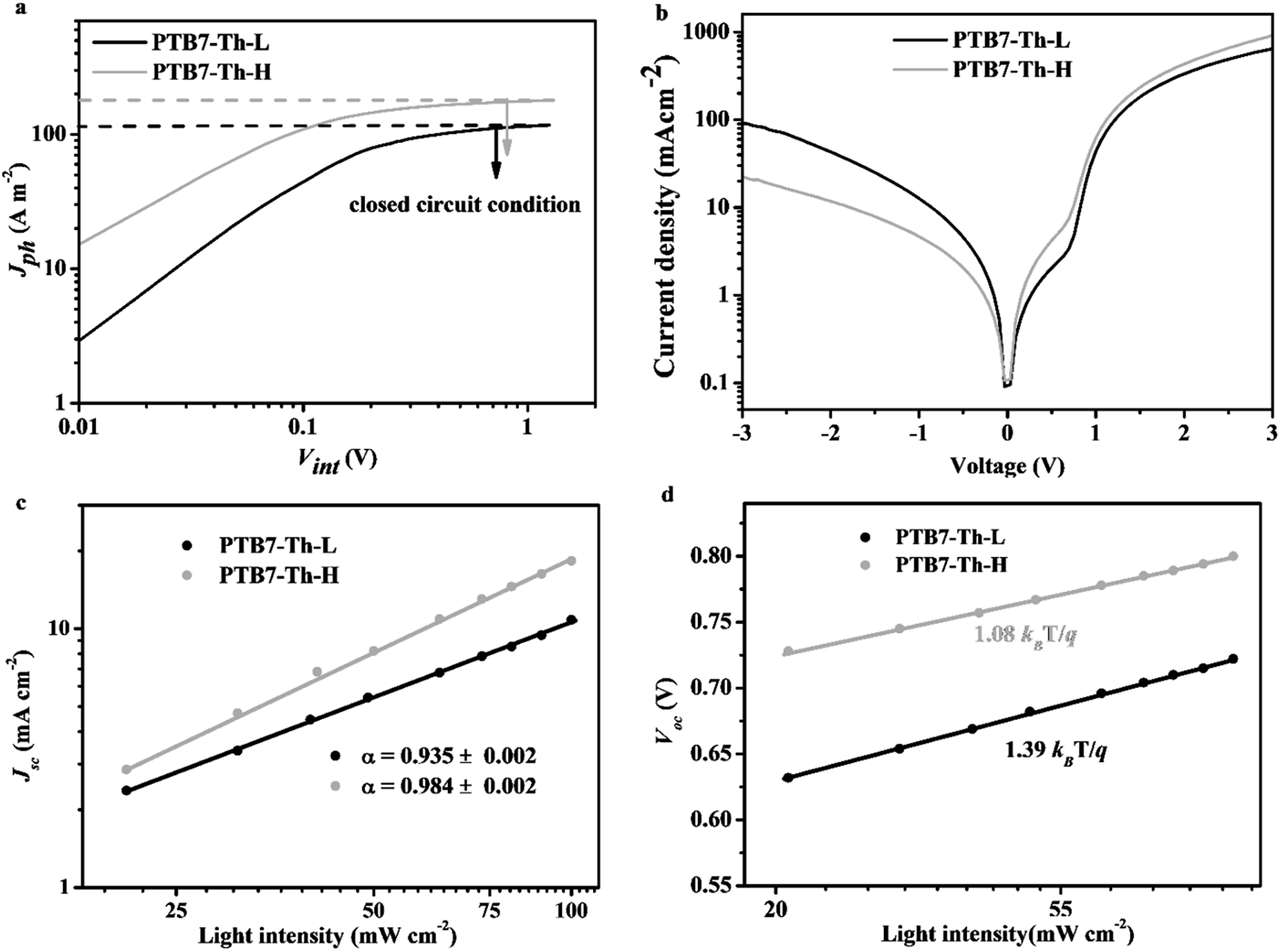 | ||
| Fig. 5 Photogenerated current density versus effective voltage curves under AM 1.5G illumination (a); J–V characteristics of the optimized BHJ devices swept from −3 V to +3 V in the dark (b); and dependence of Jsc on light intensity (c) and dependence of Voc on light intensity (d) for BHJ PSCs based on PTB7-Th-L:PC71BM and PTB7-Th-H:PC71BM blends. | ||
To gain more quantitative information on distinctive device performance, we next measured the hole (μh) and electron (μe) mobility of the polymer:PC71BM blends in hole- and electron-only devices via the space charge limited current (SCLC) method as indicated in Fig. S9.† Relative to the PTB7-Th-L:PC71BM blend with a μh of 0.25 × 10−4 cm2 V−1 s−1 and a μe of 0.86 × 10−4 cm2 V−1 s−1 (μh/μe = 0.30), the PTB7-Th-H:PC71BM blend exhibits higher charge carrier mobility with a μh of 1.52 × 10−4 cm2 V−1 s−1 and a μe of 1.21 × 10−4 cm2 V−1 s−1 (μh/μe = 1.25). Obviously, not only is the mobility of charge carriers (especially the hole mobility) improved, the transport balance of charge carriers has also been greatly improved in the PTB7-Th-H:PC71BM blend, which are beneficial for reducing space charge accumulation and thus facilitating charge transport and extraction to offer a higher Jsc and FF within the device.
We further probed the incident light intensity (Plight) dependent J–V characteristics of the PSC devices with the two polymers to investigate the state of charge recombination.53,54Fig. 5c exhibits logarithmic plots of Jsc as a function of Plight and the data were fitted according to Jsc ∝ Plightα, where a value of α close to unity is indicative of weak bimolecular recombination under closed circuit conditions. The device with PTB7-Th-H shows a relatively higher α value of 0.984 compared to 0.935 obtained from the device with PTB7-Th-L, suggesting that the bimolecular recombination loss is much smaller within the PTB7-Th-H:PC71BM blend. The state of the monomolecular recombination under open circuit conditions within both devices was examined by treating Voc as a function of Plight following Voc ∝ (nkBT/q)ln(Plight), where kB is the Boltzmann constant (1.38 × 10−23 J K−1), T is temperature in kelvin, and q is the elementary charge (1.6 × 10−19 C). It has been demonstrated that a value of n close to 1 would indicate that the bimolecular recombination dominates the recombination loss. When additional Shockley–Read–Hall (SRH) or trap-assisted recombination (regarded as monomolecular recombination) is involved, the competition with the bimolecular recombination would lead to an increased n, and the farther away the n moves from 1 (or the closer the n approaches to 2), the more serious the monomolecular recombination. As shown in Fig. 5d, the slopes for both devices were determined to be 1.08 kBT/q with an n value of 1.08 and 1.39 kBT/q with an n value of 1.39 for the devices with PTB7-Th-H and PTB7-Th-L respectively, indicating a significantly suppressed monomolecular recombination within the PTB7-Th-H:PC71BM blend under open circuit conditions.
3.6 Morphology of the BHJ blend films
Besides the improved absorption capability of PTB7-Th-H, we can find that the significantly improved Jsc and FF values are mainly attributed to enhanced photo-induced charge transfer and charge carrier generation and charge transport and suppressed charge recombination within the PTB7-Th-H based device. A variety of BHJ PSC paradigms have ignited considerable concern on the optimized morphology of a BHJ blend due to the determinant role it plays in governing each photoelectric conversion step in a broad range of length scales from the molecular scale to the nanoscale and even to the macroscale. Taking into account the similar molecular ordering of the two polymers with the preferential face-on orientation in the neat films as mentioned above, we recognized that the polymer:PC71BM blends could present a completely different microstructure in contrast to the pure polymers due to the effect of strong polymer:PC71BM phase separation.The surface morphology of the polymer:PC71BM blends was first investigated by tapping-mode atomic force microscopy (AFM) as indicated in Fig. S10.† The height images of the blend films with the two polymers exhibit very similar microstructures with small-length scales and a root-mean-square roughness of ∼5.0 nm. Similar phase separation features can be observed as well from the phase images of the two BHJ blends. Despite the fact that the domain sizes and surface features are somehow sensitive to the molecular weight of the polymer donor for a specific polymer:fullerene blend, the AFM images have limitations in resolving the structural variation between the two blends in our case. However, given the fact that there is almost complete quenching of the fluorescence within the PTB7-Th-H:PC71BM blend, we can reasonably speculate that the phase separation is more optimized than that within the PTB7-Th-L:PC71BM blend.
Further investigation of the BHJ blends by GIWAXS offers more insight into the molecular packing and orientation of crystallites within the films. Depicted in Fig. 6 is the line-cut profiles of the polymer:PC71BM blends in both in-plane and out-of-plane directions deduced from the corresponding GIWAXS patterns as shown in Fig. S11.† Upon addition of PC71BM, PTB7-Th-L exhibits (100) diffractions at ∼0.32 Å−1 with a d-spacing value of ∼19.62 Å in the OOP direction and at ∼0.28 Å−1 with a d-spacing value of 22.43 Å in the in-plane (IP) direction, suggesting the existence of polymorphs. This ordering feature can be observed as well for the PTB7-Th-H:PC71BM blend at almost the same positions of q in both the OOP and IP directions. As such, the orientation of both polymers is switched from the preferential face-on to a coexistence of face-on and edge-on from the neat films to the BHJ blends where the polymer packing is greatly disturbed by the introduction of PC71BM. However, it is important to note that a more face-on orientation of PTB7-Th-H can be observed in the PTB7-Th-H:PC71BM blend due to stronger (100) diffraction in the IP direction than that in the OOP direction. In addition, only a weak (010) π–π stacking peak can be observed at ∼1.68 Å (a d-spacing of 3.74 Å) from the BHJ blend with PTB7-Th-H along the OOP direction. Conversely, a more edge-on orientation of PTB7-Th-L can be detected in the PTB7-Th-L:PC71BM blend due to stronger (100) diffraction in the OOP direction than that in the IP direction. It is noteworthy that the diffuse diffraction at ∼1.33 Å−1 due to PC71BM aggregation is a little bit stronger in the BHJ blend with PTB7-Th-H along both the IP and OOP directions compared to the PTB7-Th-L based BHJ blend, indicating a more ordered aggregated phase of PC71BM in the device. The more ordered PC71BM phase reveals the cause for the increase in electron mobility within the PTB7-Th-H:PC71BM device. In a BHJ PSC device, charge transport in the vertical direction is usually strongly dependent on the face-on interchain π–π stacking. A more face-on orientation of conjugated backbones of the PTB7-Th-H in the BHJ blend explains well the noticeable improvement of the charge transport with suppressed charge recombination, leading to the superior photovoltaic performance with the greatly enhanced Jsc and FF in comparison with the PTB7-Th-L based device as indicated above.
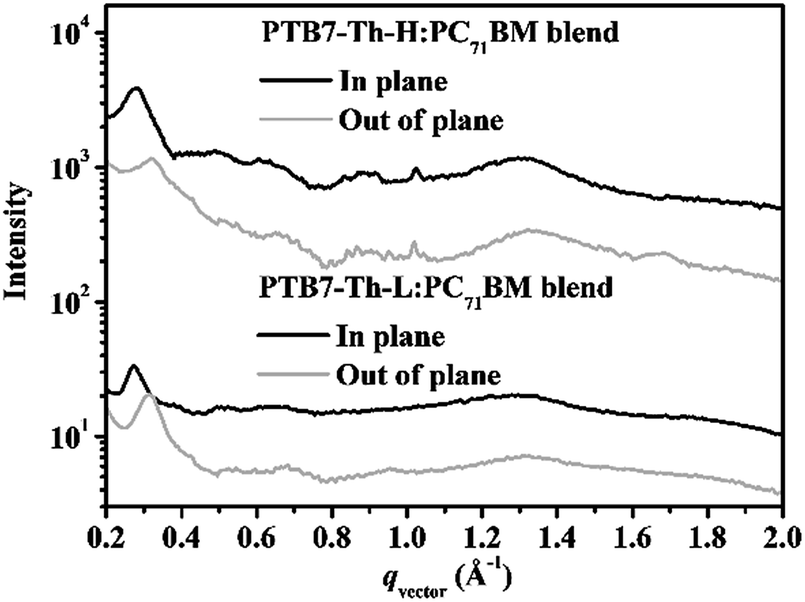 | ||
| Fig. 6 In-plane and out-of-plane linecuts of GIWAXS results of PTB7-Th-L:PC71BM and PTB7-Th-H:PC71BM blends. | ||
4. Conclusions
In this work, following the general preparation conditions for state-of-the-art PSC materials, the classic palladium catalysts of Pd2(dba)3/P(o-tol)3 and Pd(PPh3)4 were employed to produce two samples of PTB7-Th labeled PTB7-Th-L and PTB-Th-H respectively by Stille coupling copolymerization from FTT and BDTT units. The higher proportion of the amount of FTT loading into the conjugated backbone and the higher molecular weight with smaller PDI were discovered for the sample of PTB7-Th-H. Such unique structural variation relative to PTB7-Th-L endows PTB7-Th-H with strong preaggregation behavior in diluted solution and improved absorption capability both in solution and in thin film. PSCs fabricated with the PTB7-Th-H polymer demonstrated significant improvement of the PCE (8.65%) by around two times the PCE (4.07%) obtained from PTB7-Th-L. Despite similar solid state ordering of the two polymers in the neat films, the orientation of both polymers is switched from the preferential face-on to the coexistence of face-on and edge-on from the neat films to the BHJ blends. A more face-on orientation of the conjugated backbone of PTB7-Th-H in the BHJ blend contributes to an enhanced photoinduced charge carrier generation and charge transport, leading to a superior photovoltaic performance with overwhelming Jsc and FF values in comparison with the PTB7-Th-L based BHJ device. The reported findings clearly demonstrate the critical importance of choosing the right catalyst to prepare high performance D–A copolymers and preventing misreading the corresponding photovoltaic performance when an incompetent catalyst is employed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is financially supported by the National Natural Science Foundation of China (21673170) and State Key Laboratory of Advanced Technology for Materials Synthesis and Processing (2017-KF-9). X. Lu acknowledges the financial support from the RGC of Hong Kong GRF (No. 14314216) and beam time and technical support provided by the 23A SWAXS beamline at NSRRC, Hsinchu.Notes and references
- Y. Li, Acc. Chem. Res., 2012, 45, 723 CrossRef CAS PubMed.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666 CrossRef CAS PubMed.
- K. Zhang, Z. Hu, C. Sun, Z. Wu, F. Huang and Y. Cao, Chem. Mater., 2017, 29, 141 CrossRef CAS.
- Z. Hu, L. Ying, F. Huang and Y. Cao, Sci. China: Chem., 2017, 60, 571 CrossRef CAS.
- L. Dou, J. You, Z. Hong, Z. Xu, G. Li, R. A. Street and Y. Yang, Adv. Mater., 2013, 25, 6642 CrossRef CAS PubMed.
- H. Kang, G. Kim, J. Kim, S. Kwon, H. Kim and K. Lee, Adv. Mater., 2016, 28, 7821 CrossRef CAS PubMed.
- G. Luo, X. Ren, S. Zhang, H. Wu, W. C. H. Choy, Z. He and Y. Cao, Small, 2016, 12, 1547 CrossRef CAS PubMed.
- Z. Yin, J. Wei and Q. Zheng, Adv. Sci., 2016, 3, 1500362 CrossRef PubMed.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CAS.
- J. C. Hummelen, B. W. Knight, F. LePeq, F. Wudl, J. Yao and C. L. Wilkins, J. Org. Chem., 1995, 60, 532 CrossRef CAS.
- Y.-Y. Lai, Y.-J. Cheng and C.-S. Hsu, Energy Environ. Sci., 2014, 7, 1866 CAS.
- M. M. Wienk, J. M. Kroon, W. J. H. Verhees, J. Knol, J. C. Hummelen, P. A. van Hal and R. A. J. Janssen, Angew. Chem., Int. Ed., 2003, 42, 3371 CrossRef CAS PubMed.
- A. Facchetti, Mater. Today, 2013, 16, 123–132 CrossRef CAS.
- H. Benten, D. Mori, H. Ohkita and S. Ito, J. Mater. Chem. A, 2016, 4, 5340 CAS.
- Y. Liang and L. Yu, Acc. Chem. Res., 2010, 43, 1227–1236 CrossRef CAS PubMed.
- L. Ye, S. Zhang, L. Huo, M. Zhang and J. Hou, Acc. Chem. Res., 2014, 47, 1595 CrossRef CAS PubMed.
- L. Lu and L. Yu, Adv. Mater., 2014, 26, 4413–4430 CrossRef CAS PubMed.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397 CrossRef CAS PubMed.
- C. Liu, K. Wang, X. Gong and A. J. Heeger, Chem. Soc. Rev., 2016, 45, 4825 RSC.
- J.-S. Wu, S.-W. Cheng, Y.-J. Cheng and C.-S. Hsu, Chem. Soc. Rev., 2015, 44, 1113 RSC.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148 CrossRef CAS PubMed.
- B. Fan, L. Ying, Z. Wang, B. He, X.-F. Jiang, F. Huang and Y. Cao, Energy Environ. Sci., 2017, 10, 1243 CAS.
- Z. Li, X. Xu, W. Zhang, X. Meng, Z. Genene, W. Ma, W. Mammo, A. Yartsev, M. R. Andersson, R. A. J. Janssen and E. Wang, Energy Environ. Sci., 2017, 10, 2212 CAS.
- S. Xiao, Q. Zhang and W. You, Adv. Mater., 2017, 29, 1601391 CrossRef PubMed.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607 CrossRef CAS.
- J. Rivnay, S. C. B. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488 CrossRef CAS PubMed.
- Y. Cai, L. Huo and Y. Sun, Adv. Mater., 2017, 29, 1605437 CrossRef PubMed.
- L. Xue, Y. Yang, J. Xu, C. Zhang, H. Bin, Z.-G. Zhang, B. Qiu, X. Li, C. Sun, L. Gao, J. Yao, X. Chen, Y. Yang, M. Xiao and Y. Li, Adv. Mater., 2017, 29, 1703344 CrossRef PubMed.
- H. Yao, Y. Li, H. Hu, P. C. Y. Chow, S. Chen, J. Zhao, Z. Li, J. H. Carpenter, J. Y. L. Lai, G. Yang, Y. Liu, H. Lin, H. Ade and H. Yan, Adv. Energy Mater., 2017 DOI:10.1002/aenm.201701895.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 8057 CrossRef CAS.
- Q. Zhang, M. A. Kelly, N. Bauer and W. You, Acc. Chem. Res., 2017, 50, 2401 CrossRef CAS PubMed.
- W. Li, L. Yang, J. R. Tumbleston, L. Yan, H. Ade and W. You, Adv. Mater., 2014, 26, 4456 CrossRef CAS PubMed.
- Z. Ding, J. Kettle, M. Horie, S. W. Chang, G. C. Smith, A. I. Shames and E. A. Katz, J. Mater. Chem. A, 2016, 4, 7274 CAS.
- J.-H. Kim, A. Gadisa, C. Schaefer, H. Yao, B. R. Gautam, N. Balar, M. Ghasemi, I. Constantinou, F. So, B. T. O'Connor, K. Gundogdu, J. Hou and H. Ade, J. Mater. Chem. A, 2017, 5, 13176 CAS.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493 CrossRef CAS PubMed.
- T. Vangerven, P. Verstappen, J. Drijkoningen, W. Dierckx, S. Himmelberger, A. Salleo, D. Vanderzande, W. Maes and J. V. Manca, Chem. Mater., 2015, 27, 3726 CrossRef CAS.
- T. Vangerven, P. Verstappen, N. Patil, J. D'Haen, I. Cardinaletti, J. Benduhn, N. Van den Brande, M. Defour, V. Lemaur, D. Beljonne, R. Lazzaroni, B. Champagne, K. Vandewal, J. W. Andreasen, P. Adriaensens, D. W. Breiby, B. Van Mele, D. Vanderzande, W. Maes and J. Manca, Chem. Mater., 2016, 28, 9088 CrossRef CAS.
- L. Lu, T. Zheng, T. Xu, D. Zhao and L. Yu, Chem. Mater., 2015, 27, 537 CrossRef CAS.
- S.-H. Liao, H.-J. Jhuo, Y.-S. Cheng and S.-A. Chen, Adv. Mater., 2013, 25, 4766 CrossRef CAS PubMed.
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625 CrossRef CAS PubMed.
- F. Zhao, S. Dai, Y. Wu, Q. Zhang, J. Wang, L. Jiang, Q. Ling, Z. Wei, W. Ma, W. You, C. Wang and X. Zhan, Adv. Mater., 2017, 29, 1700144 CrossRef PubMed.
- Y. Jin, Z. Chen, M. Xiao, J. Peng, B. Fan, L. Ying, G. Zhang, X.-F. Jiang, Q. Yin, Z. Liang, F. Huang and Y. Cao, Adv. Energy Mater., 2017, 7, 1700944 CrossRef.
- S. Chen, Y. Liu, L. Zhang, P. C. Y. Chow, Z. Wang, G. Zhang, W. Ma and H. Yan, J. Am. Chem. Soc., 2017, 139, 6298 CrossRef CAS PubMed.
- H. Bin, L. Gao, Z.-G. Zhang, Y. Yang, Y. Zhang, C. Zhang, S. Chen, L. Xue, C. Yang, M. Xiao and Y. Li, Nat. Commun., 2016, 7, 13651 CrossRef CAS PubMed.
- K. Kawashima, T. Fukuhara, Y. Suda, Y. Suzuki, T. Koganezawa, H. Yoshida, H. Ohkita, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2016, 138, 10265 CrossRef CAS PubMed.
- Y. Liang, S. Xiao, D. Feng and L. Yu, J. Phys. Chem. C, 2008, 112, 7866 CAS.
- J. Razzell-Hollis, J. Wade, W. C. Tsoi, Y. Soon, J. Durrant and J.-S. Kim, J. Mater. Chem. A, 2014, 2, 20189 CAS.
- J. Razzell-Hollis, W. C. Tsoi and J.-S. Kim, J. Mater. Chem. C, 2013, 1, 6235 RSC.
- C. M. Proctor, M. Kuik and T.-Q. Nguyen, Prog. Polym. Sci., 2013, 38, 1941 CrossRef CAS.
- M. Yang, T.-K. Lau, S. Xiao, J. Gao, W. Wang, X. Lu, S. Zhang, J. Wu, C. Zhan and W. You, ACS Appl. Mater. Interfaces, 2017, 9, 35159 CAS.
- Y. Jiang, S. Xiao, B. Xu, C. Zhan, L. Mai, X. Lu and W. You, ACS Appl. Mater. Interfaces, 2016, 8, 11658 CAS.
- Y. Jiang, M. Yang, X. Huang, J. Gao, C. Zhan and S. Xiao, Polym. Chem., 2015, 6, 1383 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General information on materials, synthesis and structural characterization, TGA, 1H-NMR spectra, high temperature GPC chromatograms, cyclic voltammogram curves of the two PTB7-Th samples, absorption spectra of the BHJ blends, fluorescence spectra of films, charge carrier mobility testing results obtained by the SCLC method, AFM images, powder XRD profiles, and GIWAXS patterns. See DOI: 10.1039/c7ta09464g |
| This journal is © The Royal Society of Chemistry 2018 |