
Ultra-selective defect-free interfacially polymerized molecular sieve thin-film composite membranes for H2 purification†
Z.
Ali
,
F.
Pacheco
,
E.
Litwiller
,
Y.
Wang
,
Y.
Han
 and
I.
Pinnau
*
and
I.
Pinnau
*
King Abdullah University of Science and Technology, Advanced Membranes and Porous Materials Center, Division of Physical Sciences and Engineering, Thuwal 23955-6900, Saudi Arabia. E-mail: ingo.pinnau@kaust.edu.sa
First published on 10th October 2017
Abstract
Purification is a major bottleneck in generating low-cost commercial hydrogen. In this work, inexpensive high-performance H2 separating membranes were fabricated by modifying the commercially successful interfacial polymerization production method for reverse osmosis membranes. Defect-free thin-film composite membranes were formed demonstrating unprecedented mixed-gas H2/CO2 selectivity of ≈50 at 140 °C with a H2 permeance of 350 GPU, surpassing the permeance/selectivity upper bound of all known polymer membranes by a wide margin. The combination of exceptional separation performance and low manufacturing cost makes them excellent candidates for cost-effective hydrogen purification from steam cracking and similar processes.
Introduction
The transport sector consumes between 30 and 50% of global energy with demands continuing to increase annually.1 Coupled with established correlations between anthropogenic greenhouse gas emissions and global climate change, the need for energy efficient, environmentally friendly fuels is greater than ever.2 Hydrogen offers huge potential as an alternative fuel of the future due to its high energy storage capacity (119 MJ kg−1) and zero-emission combustion (produces only water).3–5 Approximately 8.5 × 1011 m3 of hydrogen – carrying 6 × 1012 MJ of energy – are produced annually, with over 90% obtained from fossil fuels (mainly natural gas and coal) and biomass or its derivatives.6 A much smaller fraction (∼8%) is produced using water electrolysis.6During steam cracking of natural gas to produce hydrogen (steam-methane reforming, SMR), methane and water are reformed to CO and H2 at ∼800 °C. The H2/CO mixture is then converted at about 350 °C into H2 and CO2. The composition of output streams can vary depending on the specific method employed. A typical SMR plant produces a H2/CO2 ratio of ∼75/20 with 5% methane and <1% of other impurities.6 Integrated Gasification Combined Cycle (IGCC) plants using biomass or coal feedstock can produce H2/CO2 ratios of ∼60/40.7 Currently about half of the globally synthesized hydrogen is used for the production of ammonia employed as a fertilizer by the Haber process, while most of the remaining half is utilized in hydrocracking i.e. breaking large hydrocarbons into smaller ones for use as fuel.8 Smaller quantities are used for the production of methanol, plastics, and pharmaceuticals, hydrogenation of oils, desulfurization of fuels, etc.8 Hydrogen production is currently growing at 10% annually, but it is estimated that availability of lower-cost hydrogen could immediately boost its use by 5- to 10-fold.9
To date, chemical separation processes have accounted for 10–15% of the global energy consumption.10 The state-of-the-art technologies for H2 purification, i.e. cryogenic distillation and pressure-swing adsorption, are extremely energy intensive, accounting for around 30% of the total plant capital and operating cost.11,12 Membrane-based H2/CO2 separation offers a potential path to reduce process costs and debottleneck H2 purification.
Table S1† lists the United States Department of Energy (USDOE) membrane performance targets for hydrogen purification from syngas mixtures.13,14 A number of materials are being considered for such separations, including inorganics such as carbon molecular sieves, zeolites, and metal membranes, as well as glassy polymers such as polybenzimidazole and polyimides with and without nanoparticles.15–25 The economic and environmental benefits of using membranes for H2/CO2 separation have been discussed by Merkel et al., suggesting that H2/CO2 selectivities greater than 10 can significantly reduce hydrogen production cost.7,14,26 Proteus™ by Membrane Technology & Research Inc. is a proprietary commercial membrane offering a H2/CO2 selectivity of approximately 11 with a H2 permeance of 500 GPU (1 GPU = 10−6 cm3 (STP) cm−2 s−1 cmHg−1) at 150 °C mixed-gas operation.14
Thin-film composite (TFC) reverse osmosis (RO) membranes constitute the most successful implementation of membrane technology in large-scale industrial separation processes due to their unmatched combination of high water flux and salt rejection. Their high water flux results from the extremely thin selective polyamide layers made by interfacial polymerization (IP). Polymers made by this process have been applied widely for industrial use, including reverse osmosis and nanofiltration membranes as well as microcapsules.27
Fig. S3† shows the structure of the partially crosslinked fully aromatic polyamide layer fabricated by reacting m-phenylenediamine (MPD) and trimesoyl chloride (TMC) on polymeric supports, pioneered by Cadotte and commercially named FT-30.28 This TFC is currently employed in more than 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 desalination plants, accounting for 90% of the global market.29 In commercial settings, FT-30-type RO membranes are produced by impregnating (via dipping or spraying) a highly porous support material (usually polysulfone) with MPD dissolved in water. Typically, excess solution is removed from the surface by using an air knife or a rubber roller. The diamine-soaked porous polysulfone support is then exposed to TMC dissolved in a hydrocarbon solvent (typically n-hexane or Isopar®) between 1 and 60 seconds.28 Most commonly, solutions in the IP process are applied at room temperature (20–25 °C). The membrane is then immediately exposed to high temperatures (∼80–100 °C) for drying and curing of the polyamide. Commercial RO membranes made by the IP process have been laboriously studied and reported in the literature with no usable gas separation properties. Gas permeation studies of dry FT-30-type RO membranes30–33 demonstrated Knudsen diffusion transport, implying the presence of mesoporous surface defects. Interestingly, Louie et al. demonstrated that plugging the surface defects by coating FT-30-type membranes with a rubbery polyether–polyamide block copolymer (PEBAX® 1657) showed some potential for H2/CO2 separation.32
000 desalination plants, accounting for 90% of the global market.29 In commercial settings, FT-30-type RO membranes are produced by impregnating (via dipping or spraying) a highly porous support material (usually polysulfone) with MPD dissolved in water. Typically, excess solution is removed from the surface by using an air knife or a rubber roller. The diamine-soaked porous polysulfone support is then exposed to TMC dissolved in a hydrocarbon solvent (typically n-hexane or Isopar®) between 1 and 60 seconds.28 Most commonly, solutions in the IP process are applied at room temperature (20–25 °C). The membrane is then immediately exposed to high temperatures (∼80–100 °C) for drying and curing of the polyamide. Commercial RO membranes made by the IP process have been laboriously studied and reported in the literature with no usable gas separation properties. Gas permeation studies of dry FT-30-type RO membranes30–33 demonstrated Knudsen diffusion transport, implying the presence of mesoporous surface defects. Interestingly, Louie et al. demonstrated that plugging the surface defects by coating FT-30-type membranes with a rubbery polyether–polyamide block copolymer (PEBAX® 1657) showed some potential for H2/CO2 separation.32
In this work, the successful fabrication of highly crosslinked, ultra-selective, defect-free MPD-TMC polyamide thin-film composite molecular sieve membranes is reported for the first time for H2/CO2 separation. Pure- and mixed-gas permeation experiments were performed across a range of temperatures up to 140 °C. The TFCs were further characterized using scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD) and Fourier transform infrared spectroscopy (FTIR). The performance of these inexpensive high-performance membranes exceeded the USDOE targets and the H2/CO2 permeance/selectivity upper bound for polymer membranes by a wide margin.
Experimental
Ultra-selective, defect-free MPD-TMC polyamide TFC membranes were fabricated by varying three parameters: (i) reaction time, (ii) TMC concentration and (iii) organic phase temperature. The materials and methods used are given in the ESI.† Porous polysulfone support layers (11.5 × 15.5 cm) were immersed in tap water for 24 hours followed by immersion in 2 wt/vol% of MPD dissolved in distilled water for 5 minutes. The support was then passed through a rubber roller to remove any excess droplets on the surface and fixed in a sealed Teflon frame. Isopar® was heated to the desired temperature and TMC was dissolved at a specific concentration. The TMC solution was then poured on the polysulfone surface, initiating the reaction. After the specified reaction time, excess solution was poured off. The membrane was immediately washed in the frame three times consecutively with 30 ml Isopar® and isopropanol. Finally, it was dried at room temperature for 48 hours and stored in a desiccator prior to testing. Table 1 lists the TFC membranes prepared in this study. The membrane designation is defined by (i) the reaction time between organic TMC and aqueous diamine phases (10–300 s); (ii) TMC concentration (0.1–10 wt/vol%); and (iii) organic phase temperature (20–100 °C). Data for at least three duplicate samples are reported for each permeation test.| Membrane | Reaction time (s) | Concentration (wt/vol%) | Temperature (°C) | m |
|---|---|---|---|---|
| FT-30 variant RO4 | Proprietary | |||
| 10s-0.1TMC-20C | 10 | 0.1 | 20 | N.M. |
| 60s-0.1TMC-20C | 60 | 0.1 | 20 | N.M. |
| 300s-0.1TMC-20C | 300 | 0.1 | 20 | 0.62 |
| 600s-0.1TMC-20C | 600 | 0.1 | 20 | N.M. |
| 300s-0.1TMC-60C | 300 | 0.1 | 60 | 0.66 |
| 300s-1TMC-60C | 300 | 1 | 60 | 0.55 |
| 300s-10TMC-60C | 300 | 10 | 60 | 0.39 |
| 300s-0.1TMC-100C | 300 | 0.1 | 100 | 0.89 |
Results and discussion
Commercially produced dry FT-30 membranes are known to contain micropores larger than the dimensions of gas molecules, as Knudsen selectivity has been measured in a variety of FT-30-type products.32,33 Although specific production information is proprietary, it is widely known that these membranes are made with IP reaction times under one minute. Fig. 1(a) and (b) show how defect-free polyamide layer characteristics, as indicated by significantly increased selectivity, start to emerge at longer reaction times. Permeance for H2 and He decreased 10-fold, whereas an average decrease of at least 100-fold was observed for larger gases as the reaction time was increased from 10 to 300 s. Longer reaction times presumably allow more MPD to penetrate into the reaction zone forming additional polyamide and thus closing any defects in the ultrathin-film by a diffusion-driven self-healing process, as schematically shown in Fig. S4.† During this process, the mode of transport shifted from Knudsen flow to solution/diffusion, and gas pair selectivity increased at 60 s while reaching an optimum at a reaction time of 300 s. Numerical performance data are summarised in Tables S2 and S3.† The 10s-0.1TMC-20C membrane demonstrated identical gas permeation properties to a commercial FT-30-type membrane (Sepro RO-4) and was used as the reference for comparing the performance of other TFCs in this work.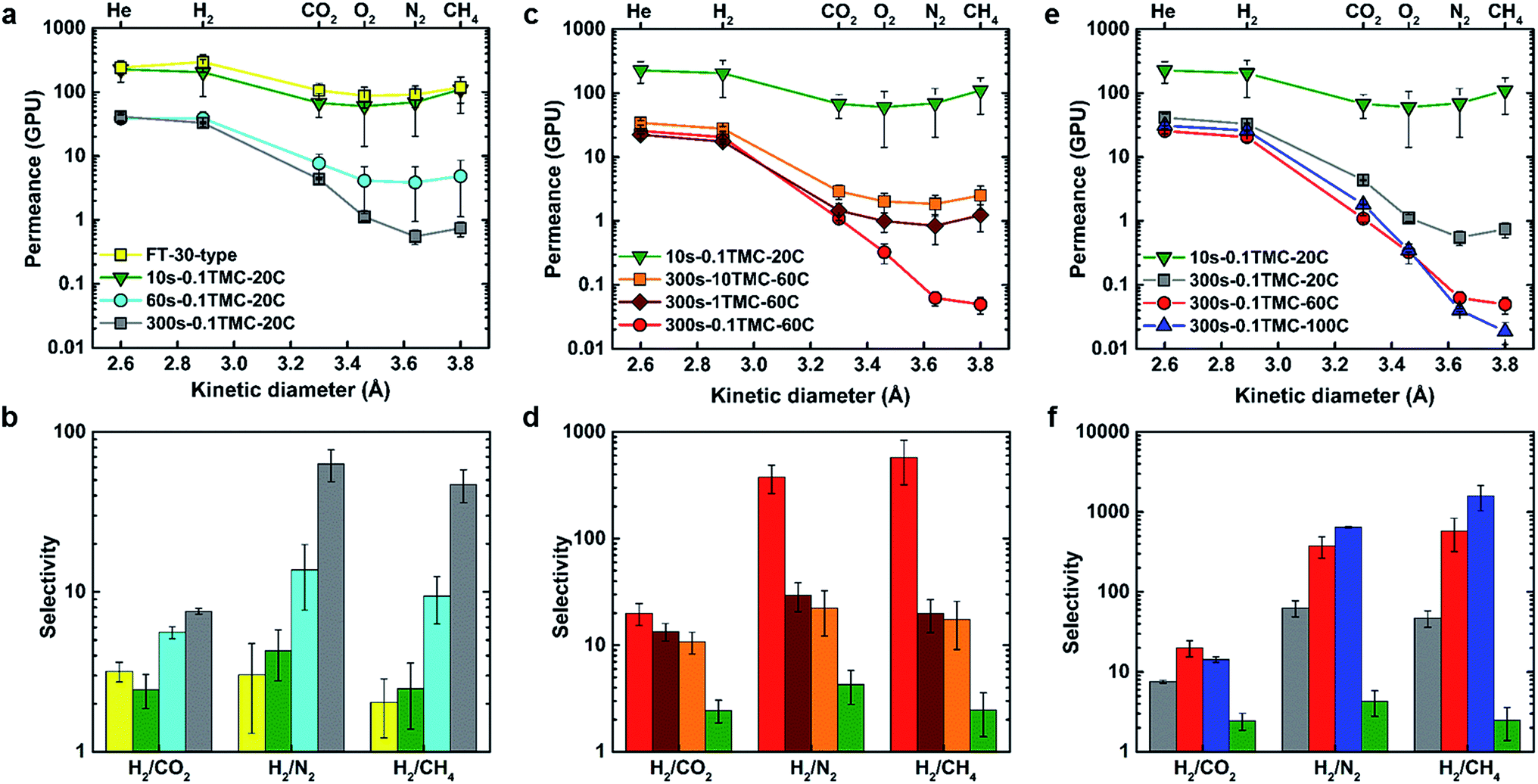 | ||
| Fig. 1 Pure-gas separation performance of polyamide thin-film composite membranes. Effect of (a and b) reaction time; (c and d) TMC concentration; and (e and f) organic phase temperature on permeance and selectivity, respectively. | ||
Fig. 1(c) and (e) show the effects of varying TMC concentration and organic phase temperature on gas permeance properties of the TFCs. No significant variation in permeance was observed for helium (kinetic diameter (kd) = 2.60 Å) and hydrogen (kd = 2.89 Å) but a clear trend of decreasing permeance started to emerge for gases with kd values larger than 3 Å (O2, CO2, N2, and CH4).34 FTIR spectra (Fig. S5†) demonstrated no visible difference in polyamide chemistry for different fabricated samples compared to the 10s-0.1TMC-20C reference membrane. However, as TMC concentration decreased, the ratio of amine to acyl chloride functional groups increased as demonstrated by XPS analysis, indicating an increase in the degree of crosslinking (Table 1). This resulted in tightening of the polyamide network, consequently hindering the transport of larger gas molecules while no significant effect was observed for smaller gases (He and H2), which resulted in a significant boost in selectivity. Similarly, increasing the organic-phase temperature also increased the degree of crosslinking, which lowered the permeance of gases larger than H2 thereby significantly enhancing selectivity. Presumably, the increase in reaction-zone temperature increased the overall reaction rate as well as solubility and diffusivity of MPD in the organic phase (reaction-zone), resulting in increased formation of amide linkages and, hence, increased crosslinking.35
Fig. 1(d) and (f) show the membrane performance results expressed in terms of selectivity. High selectivity for H2/CO2 and negligible selectivity for He/H2 imply a primary molecular-sieve-like cut-off at around 3 Å. This is clearly displayed in the XRD spectrum of the MPD-TMC powder in Fig. S6,† showing a main amorphous peak with an average chain d-spacing centered around 3.5 Å. As m increased from 0.39 to 0.66, the selectivity of hydrogen over CO2, O2, N2 and CH4 increased, implying increased ultramicroporosity. As the degree of crosslinking increased further from 0.66 to 0.89, N2 and CH4 permeance decreased (kd values for N2 and CH4 are 3.64 Å and 3.80 Å, respectively) but CO2 and O2 permeances remained unaffected. As a direct consequence, O2/N2, CO2/N2 and CO2/CH4 selectivities increased (Fig. S7†). These are all significant industrial gas separation applications for implementation of membrane technology.
SEM images, Fig. S8(a–l) and S9(a–d),† illustrate that the TFCs of this study have typical average visual polyamide ridge-and-valley-based film thicknesses of approximately 100–300 nm.36 However, it has been suggested that there is an appreciable difference between the observed average cross-sectional thickness and the actual effective thickness of the selective layer. The apparent visual thickness has been conventionally considered the true thickness of the polyamide barrier layer;35,37 however, more recent research has indicated that the effective thickness of the separation layer lies around the order of only 10–20 nm.36,38,39 The cross-sections of the defective FT-30-type reference membrane (10s-0.1TMC-20C) and the defect-free, highly gas-selective polyamide TFC of this work (300s-0.1TMC-100C) are shown in Fig. 2. Although it is difficult to clearly assign a thickness to the ultrathin selective polyamide barrier layer of both membrane types, it is clear that the PA layer is thicker and more tightly packed in the membrane made with both longer reaction time and higher reaction temperature (Fig. 2(b)).
 | ||
| Fig. 2 SEM cross section of (a) 10s-0.1TMC-20 and (b) 300–0.1TMC-100C highlighting the variation of morphology for lowest and highest performing samples, respectively. | ||
Gas permeation data for an aromatic network polyamide based on MPD and TMC are not available because isotropic films cannot be produced due to the inherent insolubility of the crosslinked polymer. However, Weinkauf, Kim and Paul40 reported the gas permeation properties of thick films of a related linear, amorphous aromatic polyamide made from phenylenediamine isomers and terephthaloyl chloride. The isotropic poly(phenylene terephthalamide) films used in their study were ∼25–100 μm thick and demonstrated gas barrier behavior with O2 and CO2 permeabilities at 35 °C of only 0.026 and 0.1 Barrer, respectively (1 Barrer = 1 × 10−10 cm3 (STP) cm cm−2 s−1 cmHg−1). Because of their extremely low gas permeabilities, aromatic polyamides have been rarely considered for membrane-based gas separation processes.41 However, as demonstrated here, this disadvantage can be overcome by fabricating ultra-thin films allowing the exploitation of highly selective barrier materials with industrially useable performance characteristics.
Pure- and mixed-gas temperature dependence
The membranes fabricated in this study showed excellent potential for syngas separations at 22 °C. However, equally important requirements for membranes in H2 purification from syngas are stability at high feed temperatures (∼150–250 °C) and high pressures (>7 bar).Fig. 3(a) shows the performance of the 300s-0.1TMC-100C membrane as a function of temperature using pure-gas H2 and CO2 measurements. Permeance for both gases showed excellent Arrhenius regression with temperature (ESI Section 11†). H2 experienced a larger increase in permeance compared to CO2 presumably due to reduced sorption of CO2 at higher temperatures. At 140 °C, H2 permeance increased to 275 ± 4 GPU with a H2/CO2 selectivity of 95.5 ± 5, the highest reported pure-gas selectivity to date for any polymer membrane. Activation energies for H2 and CO2 were calculated to be 8.50 and 1.20 kJ mol−1, respectively. The surprisingly lower activation energy of permeation for CO2 than H2 was previously observed by Li et al. for a series of polybenzimidazoles.23 It was suggested that the smaller relative increase in CO2 permeability with temperature resulted from strong CO2–polymer interactions. Weinkauf et al. observed a similar trend for aromatic polyamides and evidenced strong polymer/CO2 interactions by large negative CO2 heat of sorption values.40
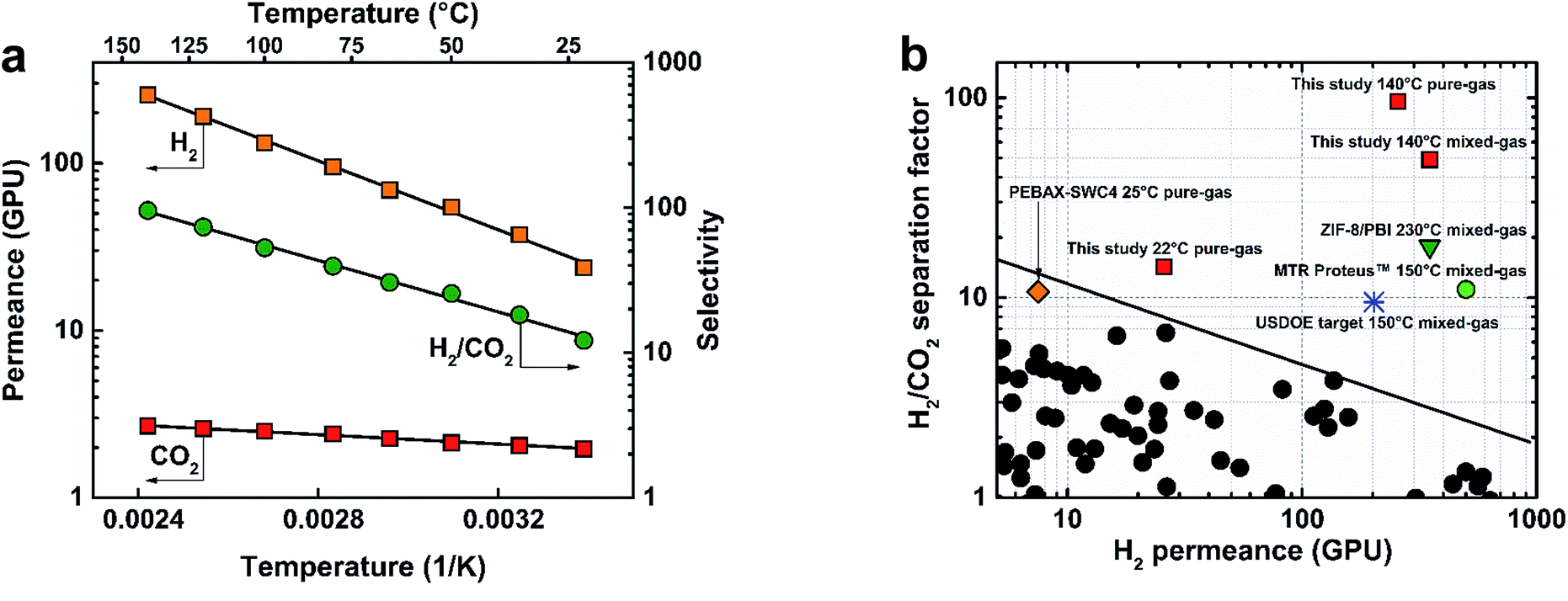 | ||
| Fig. 3 (a) TFC membrane (300s-0.1TMC-100C) pure-gas temperature dependence for H2 and CO2, and (b) Robeson plot for performance comparison of the TFC membrane studied here (300s-0.1TMC-100C) with other membrane types. Permeance/selectivity upper bound adapted from Robeson (2008) assuming 1 μm-thick films.42 USDOE target membrane requirements for H2/CO2 separation13,14 and performance data for MTR Proteus™,14 ZIF-8/PBI,19 and PEBAX-coated SWC4 (PEBAX-SWC4).32 | ||
Mixed-gas separation was conducted using a 1:1 H2/CO2 feed at 140 °C to provide more realistic performance data in industrial systems. Fig. 3(b) shows pure- and mixed-gas data for the 300s-0.1TMC-100C membrane compared to state-of-the-art polymer membranes in the permeance/selectivity plot for H2/CO2 separation.42 An average stabilized H2 permeate concentration of 98% was achieved, translating to an unprecedented mixed-gas separation factor of 50 ± 4 with a hydrogen permeance of 350 ± 15 GPU at 140 °C. Our mixed-gas permeation results clearly demonstrated unparalleled performance of the defect-free polyamide TFCs for H2/CO2 separation with properties far exceeding those of all state-of-the-art polymer membranes when tested under industrially relevant conditions. It is important to note that the aromatic polyamide is thermally stable up to ∼300 °C (Fig. S10†). However, the upper operational temperature of the TFC membranes reported here is limited by the thermal stability of the porous polysulfone support. Hence, future developments of TFC membranes for high-temperature H2/CO2 separation (∼200–300 °C) must be directed towards the development of a more thermally stable porous support.
Conclusions
The growing need for cleaner energy has dramatically increased interest in separations using membrane systems. Highly crosslinked, ultra-selective, defect-free MPD-TMC membranes were successfully fabricated in this study showing tremendous potential for H2/CO2 separation in syngas applications as well as a number of other challenging gas separations. These membranes exhibited unprecedented H2/CO2 selectivity combined with very high H2 permeance at 140 °C, surpassing the performance of all other reported polymer membranes to date.Fortuitously, these ultra-high-performance membranes can be produced by making only small changes to existing commercial membrane manufacturing processes by interfacial polymerization. Therefore, their fabrication cost should be similar to those of standard RO membranes of only ≈1–2 $ per ft2,43 which would lower the membrane cost by 50–100-fold based on the DOE target value of 100 $ per ft2. This study demonstrated that varying fabrication parameters can tune permselectivity to meet the needs of specific processes. A few simple modifications to a time-tested commercial membrane fabrication process can produce membranes that meet a key industrial need. With rapidly developing economic and environmental pressures to increase efficiencies for separation processes, such highly selective, low-cost, commercial barrier materials fabricated as ultra-thin films show potential for a paradigm shift to streamline the industrial use of membranes for hydrogen separations.
TFCs based on MPD and TMC also demonstrated remarkable selectivity for O2/N2, CO2/CH4, H2/N2 and CO2/N2 separations. However, the most promising TFC membrane for H2/CO2 separation (300s-0.1TMC-100C) exhibited very low CO2 and O2 permeances of only 1.8 and 0.4 GPU, respectively. Recent work by Tsai et al. demonstrated more promising results for O2/N2 separation for interfacially polymerized TFCs made by the reaction of TMC with piperazine.44
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. P. Lively and D. S. Sholl, Nat. Mater., 2017, 16, 276–279 CrossRef CAS PubMed.
- J. Hansen, R. Ruedy, M. Sato and K. Lo, Rev. Geophys., 2010, 48, RG4004 CrossRef.
- D. Teichmann, W. Arlt and P. Wasserscheid, Int. J. Hydrogen Energy, 2012, 37, 18118–18132 CrossRef CAS.
- United States Congress, Energy Policy Act: Public Law 102-486, Washington, 1992 Search PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- N. W. Ockwig and T. M. Nenoff, Chem. Rev., 2007, 107, 4078–4110 CrossRef CAS PubMed.
- T. C. Merkel, M. Zhou and R. W. Baker, J. Membr. Sci., 2012, 389, 441–450 CrossRef CAS.
- P. Häussinger, R. Lohmüller and A. M. Watson, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011 Search PubMed.
- Y. Bilge, C. Guenter, M. C. Petri and C. Forsberg, Configuration and Technology Implications of Potential Nuclear Hydrogen System Applications Prepared by Nuclear Engineering Division, Chicago, 2005 Search PubMed.
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 6–8 CrossRef PubMed.
- T. L. Buchanan, M. G. Klett and R. L. Schoff, Capital and Operating Cost of Hydrogen Production from Coal Gasification, Pittsburgh, 2003 Search PubMed.
- R. Spillman, in Membrane Science and Technology, ed. R. D. Noble and S. A. Stern, Elsevier B.V., 2nd edn, 1995, vol. 2, pp. 589–667 Search PubMed.
- United States Department of Energy, Basic Research Needs for the Hydrogen Economy, Workshop on hydrogen production, storage and use, Lemont, 2003 Search PubMed.
- United States Department of Energy, Advanced Carbon Dioxide Capture R&D Program: Technology Update: Pre-combustion Membranes, Pittsburgh, 2013 Search PubMed.
- K. J. Bryden and J. Y. Ying, J. Membr. Sci., 2002, 203, 29–42 CrossRef CAS.
- Y. H. Ma, Adv. Membr. Technol. Appl., 2008, 671–684 CAS.
- D. R. Pesiri, B. Jorgensen and R. C. Dye, J. Membr. Sci., 2003, 218, 11–18 CrossRef CAS.
- K. A. Berchtold, R. P. Singh, J. S. Young and K. W. Dudeck, J. Membr. Sci., 2012, 415–416, 265–270 CrossRef CAS.
- T. Yang and T.-S. Chung, Int. J. Hydrogen Energy, 2013, 38, 229–239 CrossRef CAS.
- S. S. Hosseini, N. Peng and T. S. Chung, J. Membr. Sci., 2010, 349, 156–166 CrossRef CAS.
- S. Japip, K. S. Liao and T. S. Chung, Adv. Mater., 2017, 29, 1–6 CrossRef PubMed.
- P. Li, Z. Wang, Z. Qiao, Y. Liu, X. Cao, W. Li, J. Wang and S. Wang, J. Membr. Sci., 2015, 495, 130–168 CrossRef CAS.
- X. Li, R. P. Singh, K. W. Dudeck, K. A. Berchtold and B. C. Benicewicz, J. Membr. Sci., 2014, 461, 59–68 CrossRef CAS.
- S. C. Kumbharkar, Y. Liu and K. Li, J. Membr. Sci., 2011, 375, 231–240 CrossRef CAS.
- S. C. Kumbharkar and K. Li, J. Membr. Sci., 2012, 415–416, 793–800 CrossRef CAS.
- United States Department of Energy, Cost and Performance Baseline for Fossil Energy Plants: Report Number DOE/NETL-2007/1281, Pittsburgh, 2007, vol. 4 Search PubMed.
- M. J. T. Raaijmakers and N. E. Benes, Prog. Polym. Sci., 2016, 63, 86–142 CrossRef CAS.
- J. E. Cadotte, US Pat., 4,277,344, 1981.
- S. S. Shenvi, A. M. Isloor and A. F. Ismail, Desalination, 2015, 368, 10–26 CrossRef CAS.
- J. S. Louie, I. Pinnau and M. Reinhard, J. Membr. Sci., 2011, 367, 249–255 CrossRef CAS.
- J. Duan, PhD thesis, King Abdullah University of Science and Technology, 2014.
- J. S. Louie, I. Pinnau and M. Reinhard, J. Membr. Sci., 2008, 325, 793–800 CrossRef CAS.
- J. Albo, J. Wang and T. Tsuru, J. Membr. Sci., 2014, 449, 109–118 CrossRef CAS.
- N. Mehio, S. Dai and D. E. Jiang, J. Phys. Chem. A, 2014, 118, 1150–1154 CrossRef CAS PubMed.
- A. K. Ghosh, B. H. Jeong, X. Huang and E. M. V Hoek, J. Membr. Sci., 2008, 311, 34–45 CrossRef CAS.
- F. Pacheco, R. Sougrat, M. Reinhard, J. O. Leckie and I. Pinnau, J. Membr. Sci., 2016, 501, 33–44 CrossRef CAS.
- L. Lin, C. Feng, R. Lopez and O. Coronell, J. Membr. Sci., 2016, 498, 167–179 CrossRef CAS.
- F. A. Pacheco, I. Pinnau, M. Reinhard and J. O. Leckie, J. Membr. Sci., 2010, 358, 51–59 CrossRef CAS.
- S. Karan, Z. Jiang and A. G. Livingston, Science, 2015, 348, 1347–1351 CrossRef CAS PubMed.
- D. H. Weinkauf, H. D. Kim and D. R. Paul, Macromolecules, 1992, 25, 788–796 CrossRef CAS.
- H. H. Hoehn and J. W. Richter, US Pat., 3,899,309, 1975.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- X. Chen, Z. Zhang, L. Liu, R. Cheng, L. Shi and X. Zheng, Desalination, 2016, 397, 185–193 CrossRef CAS.
- C.-W. Tsai, C. Tsai, R.-C. Ruaan, C.-C. Hu and K.-R. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 5563–5568 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, methods, FTIR, XPS, XRD, TGA, FESEM, and gas permeation data. See DOI: 10.1039/c7ta07819f |
| This journal is © The Royal Society of Chemistry 2018 |