
Experimental screening of perovskite oxides as efficient redox materials for solar thermochemical CO2 conversion†
Mahesh M.
Nair
and
Stéphane
Abanades
 *
*
Processes, Materials and Solar Energy Laboratory, PROMES-CNRS UPR 8521, 7 rue du Four Solaire, 66120 Font Romeu, France. E-mail: Stephane.Abanades@promes.cnrs.fr
First published on 5th February 2018
Abstract
Developing efficient redox materials for the thermochemical synthesis of renewable fuels from CO2 and H2O via two-step redox cycling by utilizing concentrated solar energy is a major challenge that needs to be overcome for the realization of the solar fuel technology. Metal oxide redox materials are receiving recent interest on this regard, among which perovskite structured mixed oxides with lattice oxygen transfer are particularly important. In this contribution, we examined a series of perovskites and related Ruddlesden–Popper (RP) structures for their effectiveness in solar thermochemical conversion of CO2. All the materials used in this study were synthesized from the corresponding oxides by following the conventional solid state method. Substitutions were made on the cationic A and B sites in the structure in order to modify their redox properties and to enhance their stability under the reaction conditions. In conjugation, with the compositional tailoring of the perovskite materials, the temperatures of reduction and re-oxidation were varied. Issues regarding carbonate formation were identified, for some compositions so as to affect their CO2-splitting efficiency. Also in some cases, despite the higher extent of reduction observed at low temperatures, given the presence of Co4+, Fe4+ valence states, the extent of re-oxidation and stability were comparatively lower (case of (La,Sr)(Co,Fe)O3, (Ba,Sr)(Co,Fe)O3). The best performance among the investigated series was observed for La(B,B′)O3 perovskites with Mn and Ni or Mn and Co together in the B site, as well as strontium doped lanthanum manganites with perovskite (La0.5Sr0.5MnO3) and parent RP structures. Ruddlesden–Popper materials with only Mn occupying the B-site (LaSrMnO4, LaSr2Mn2O7), exhibited noteworthy thermochemical stability for two-step redox cycling and the amount of produced CO was consistently higher without any microstructural or morphological optimization at this stage.
Introduction
Thermochemical splitting of H2O/CO2 using concentrated solar energy has recently emerged as a promising strategy for producing alternative clean fuels. Particularly, the conversion of CO2 into fuels puts forward an additional advantage that a greenhouse gas is being effectively utilized and up-graded by solar energy, instead of being a waste with a disposal cost. The solar energy resource is intermittent, diffuse and unevenly distributed and its storage in the form of solar fuels is essential for the transition away from fossil fuels. Here, the efficiency of the process relies on the redox properties of the materials employed for carrying out the conversion. Several volatile and non-volatile metal oxides were previously studied for this purpose, including ZnO, ferrites, CeO2, etc.1–11 Perovskites are one recent option for applications as redox materials in solar fuel production.12–27 The physicochemical properties of these materials can be tailored owing to their ability to host a variety of metal cations in the A and B site of the structure; either partially or completely.28–32 The resulting anion non-stoichiometry and formation of cations in unusual oxidation states could strongly influence the redox performance of these materials.In general, the two-step thermochemical dissociation of CO2/H2O over perovskites can be represented as follows:
| ABO3 → ABO3−δ + δ/2O2 | (1) |
| ABO3−δ + δCO2 (H2O) → ABO3 + δCO (H2) | (2) |
In the first oxygen producing endothermic step (eqn (1)), perovskite is partially reduced to a non-stoichiometric oxide using high-temperature solar heat. In the subsequent low temperature exothermic step (eqn (2)), the reduced perovskite is oxidized with H2O and/or CO2 producing H2 and/or CO, along with the stoichiometric oxide. The global process thus valorises CO2 using concentrated solar energy as a source of clean, non-polluting, high-temperature heat, and produces syngas (mixture of H2 and CO) that is the building block for a wide variety of synthetic fuels. The produced CO can be used to synthesize H2 from the water–gas shift reaction or further processed via a Fischer–Tropsch synthesis to produce synthetic liquid hydrocarbons as convenient combustible fuel for the transportation sector. Physicochemical stability of the redox materials under the operating conditions without hindering the performance is an important prerequisite for thermochemical solar fuel production. However cation segregation and high temperature sintering causes major concern as far as perovskites are concerned.27 Additionally, exposure to CO2 may induce the formation of carbonates especially in the presence of alkaline earth metals such as Sr, Ca or Ba in the structure.27,28 Such carbonate formation could seriously hinder the performance of these materials. Therefore further screening to deduce the optimum perovskite compositions in accordance with the aforementioned facts is necessary for the advancement of solar thermochemical fuel production.
Starting from the initial studies by Scheffe et al.,12 and McDaniel et al.,13 a variety of authors investigated perovskite structured oxides for fuel production under solar thermal conditions.14–26 Significant fundamental insights regarding the redox functioning of these materials were obtained from these studies. For example, partial substitution of the A site with Sr was found to increase the oxygen vacancy concentration as well as the fraction of B-cations with higher oxidation states, which in turn modified the thermodynamics and kinetics of the redox reactions.12,13 Demont et al., studied the redox performance of a series of perovskites along with their parent Ruddlesden–Popper structures for H2 production and correlated the performance with their crystal chemistries.14 Further, Bork et al., showed that the simultaneous inclusion of Cr and Co in the B site of the structure could significantly lower the reaction temperatures by up to 300 °C.18 More recent studies performed in our group demonstrated that the method of synthesis can critically influence the redox performance of perovskites. In particular, La0.5Sr0.5MnO3 synthesized by the Pechini method exhibited optimal performance for the solar thermochemical splitting of CO2. Our studies also indicated that the presence of promotional agents such as MgO can influence the efficiency of these materials to a small extent.25 For instance, the addition of MgO resulting in the formation of composites was used to limit the diffusion at the grain boundaries of perovskites in order to stabilize the structure and improve the thermochemical performance, thanks to the beneficial effect of MgO addition for limiting the grain size growth.25
Research studies currently aim to develop innovative and robust redox materials that can withstand the severe operating conditions of the solar thermochemical process (high-temperature, reducing/oxidizing atmospheres), and that feature high fuel production capabilities at reasonable reaction rates (to maintain the reaction duration as low as possible) with minimum temperature gap between the redox steps (to reduce the thermal losses). The reaction reversibility, oxygen exchange capacity of the materials and thermochemical fuel production yield and stability need to be experimentally investigated, especially in the field of perovskites because the number of suitable candidate materials still remains restricted. Our screening study identifies potentially attractive materials based on perovskites for the CO2 conversion application and further eliminates some formulations that exhibit poor CO2-splitting activity.
In the present work, we synthesized a series of perovskite and Ruddlesden–Popper structured oxide materials by the solid-state method and investigated the physicochemical changes taking place in these materials during thermochemical CO2 splitting. Doping A and B sites with a series of alkaline earth and transition metal cations respectively, was carried out to evaluate the variations in the performance of these materials with emphasis on efficiency and stability. The study thus encompasses a large range of materials formulations for comparing their CO2 splitting activity under similar experimental conditions and procedure. This allows identifying the most efficient materials for the targeted application. Structural characterizations were performed using wide angle XRD. Then, thermochemical reactivity and CO2-splitting efficacy during high-temperature redox cycling were evaluated using TGA. Further, the effects of temperature variations and carbonate formation on the performance of these materials were monitored. Different classes of mixed ionic-electronic conducting materials were considered encompassing: (1) La(B,B′)O3 (with B = Mn, Fe and B′ = Co, Ni) in which the doping with Mn or Fe cations favours the oxidation of LaCoO3 and LaNiO3; (2) La1−xSrx(Co,Fe)O3 and Ba1−xSrx(Co,Fe)O3 for the stabilization of Co4+, Fe4+ which favours reduction at low temperature; (3) La1−xSrxMnO3 in which substitution of La3+ by Sr2+ is used for controlling the Mn4+/Mn3+ ratio; and (4) LaSrMO4 and LaSr2M2O7 Ruddlesden–Popper phases (perovskite parent structures). Most of the materials studied here stand out by their larger thermal reduction capabilities and ability to form oxygen vacancies at modest temperatures in the range 1000–1400 °C when compared to reference non-stoichiometric compounds such as spinel ferrites or fluorite-structured ceria-based materials.
Experimental
Material synthesis
Polycrystalline perovskite and Ruddlesden–Popper (RP) structured mixed metal oxides were synthesized by following solid-state method. Binary oxides and carbonates were used as precursors in the required stoichiometry. BaCO3, SrCO3, Co3O4, NiO, Fe2O3, and MnO2 were used as received (99.99% purity). La2O3 was fired at 950 °C for 6 h prior to being weighted, in order to decompose the contaminant La(OH)3. Weighed precursors in desired proportions were ground well and subjected to an initial calcination of 1000 °C. Later, an additional calcination was performed at appropriate higher temperature necessary for the desired phase formation and structure stabilization before cycling experiments. The perovskite structures were thus obtained after this final calcination step for several hours (10–16 hours of calcination for completing the solid-state synthesis). The formation of the desired final structure depends on initial precursor stoichiometry and calcination temperatures appropriate for the crystallization of the oxide. The calcination temperature was generally higher than or equal to the reduction temperature. It must be above or at least equal to the reduction temperature in order to avoid further modification of the materials structure during the high-temperature process. The temperatures were selected on the basis of the thermal stability and melting point of the materials.Characterization and thermochemical activity measurements
X-ray diffraction (XRD) was performed on a Philips PW 1820 diffractometer with a Cu-Kα radiation (αCu = 0.15418 nm, angular range = 20–100 2θ, steps = 0.02 2θ, recording time = 2 s). Identification of crystalline phases was performed by comparison with standard diffraction patterns (powder diffraction file PDF-2, International Centre for Diffraction Data, ICDD). XRD was also used to validate the possible secondary phase formation (carbonates) during the redox cycles. Morphology and surface structure were studied using a high resolution field emission scanning electron microscope (FESEM, Hitachi S-4800).High temperature reduction and re-oxidation properties were determined by performing thermogravimetric (TG) analyses to assess the oxygen exchange capacity from sample mass variations, providing a definite value of the level of reduction/re-oxidation achieved. All the redox experiments were performed using a SETARAM SETSYS Evolution device (0.03 μg resolution) equipped with a platinum crucible to monitor continuously the sample mass as a function of temperature and atmosphere. The balance (fitted with a beam articulated on a torsion ribbon) displays high stability and sensitivity (with a typical mass drift of only 0.1 μg h−1) thanks to a high-performance, optical and electronic detection fitting. The intrinsic uncertainty could be considered minor when compared with the reduction capabilities of the samples. Indeed for cycles lasting around 7 hours, the uncertainty on the mass change is weak (below 1 μg) compared to the mass variation of the samples. Blank run correction (using empty crucible) was done to eliminate the small baseline drift during non-isothermal heating caused by thermal gas expansion and buoyancy effects on the crucible.
Sample masses of approximately 120 mg were used for each analysis. Thermal reduction step was performed under an Ar flow of 0.020 Nl min−1 (99.999% purity, <2 ppm O2) at appropriate temperatures (1000–1400 °C) during 45 min of temperature plateau. Re-oxidation of the partially reduced oxides was performed under CO2 (50% in Ar) at selected temperatures (800–1050 °C) during 45 or 60 min. All heating and cooling steps were performed with a ramp rate of 20 °C min−1. The O2 release during reduction induced a mass loss (Δmred), whereas O atom reincorporation during oxidation induced a mass increase (Δmox). These mass variations were used to determine the mole amounts of O2 released during thermal reduction (nO2 = Δmred/MO2), and CO produced during oxidation (nCO = Δmox/MO).
For each cycle, the amount of oxygen lost during the reduction was associated with the amount of CO produced in the subsequent step. This is the basis for the materials performance evaluation in each cycle, which is of course dependent on the reaction duration, but the goal was here to assess the re-oxidation potential for a given material. Besides, the re-oxidation potential is not only based on the oxygen released during previous reduction step, but rather on the total amount of oxygen lost from the first cycle (the maximum reduction state reached during the whole cycle is thus taken into account). In order to ensure consistency between the first and second cycle, the reported re-oxidation yields (nCO/2nO2) were calculated based not only on the amount of O2 released during the previous reduction step, but based on the total amount of oxygen lost and re-incorporated from the first cycle.
Results and discussion
Lanthanum-based perovskites, La(B,B′)O3
A series of B-site substituted La-based perovskites synthesized by the solid-state method were initially studied for solar thermochemical splitting of CO2. In order to vary the thermodynamic redox properties by B-site tuning, several unsubstituted simple perovskite formulations were considered initially encompassing LaCoO3, LaFeO3, LaMnO3 and LaNiO3. On the basis of their free enthalpies of reduction, the thermal reduction of LaFeO3 and LaMnO3 is not favoured below 1500 °C, whereas their re-oxidation with CO2 is favoured. Inversely, the thermal reduction of LaCoO3 and LaNiO3 is favoured at lower temperatures, but re-oxidation with CO2 is not favoured. The addition of Mn or Fe in the B site is thus expected to favour the oxidation of LaCoO3 and LaNiO3. In order to outline a trade-off of the suitable redox properties for thermochemical CO2 splitting, perovskites formulations of the class LaBxB′1−xO3 were synthesized, with B = Fe, Mn and B′ = Co, Ni. Thus, Co, Fe, Mn and Ni were used as B-site cations in the perovskite structure, among which the simplest one being un-substituted LaCoO3 (LCO) synthesized by the solid-state method at 1300 °C. Composition and purity of these materials were determined by performing wide angle XRD analysis. Highly crystalline peaks corresponding to the perovskite structure with rhombohedral symmetry were observed for the materials. In the case of B-site substituted perovskites, intense peaks corresponding to the perovskite structure alone was observed for LaMn0.5Co0.5O3 (LMCO) and LaFe0.75Co0.25O3 (LFCO) calcined at 1500 °C. However under the similar calcination conditions (1500 °C), a small amount of binary oxide impurities was also observed in some cases. In this latter case, phase pure materials were not obtained even after performing calcinations for 20 h indicating that these compositions are difficult to achieve by the solid-state route. The obtained XRD profiles along with their LeBail fits are compiled in Fig. ESI-1.† Information regarding the morphology and surface structure were obtained by performing SEM analysis and the representative images are shown in Fig. ESI-2.† No significant variations were observed with respect to the changes in compositions. Agglomerated dense particles with uneven morphologies were obtained.The samples were calcined and stabilized at high temperature prior each cycling experiment. The surface area was dramatically decreased (below 1 m2 g−1) after the calcination step for all the materials during the synthesis. As a result, the major part of the surface area reduction occurred during the synthesis and the samples were thermally stabilized before the cycles. Thus, the powder was already sintered before the first cycle and further sintering during the next reduction steps had negligible effect on the surface area. Consequently, the decrease of specific surface area and the particle growth (coarsening) induced by sintering leading to longer diffusion lengths are not characteristic parameters in such high-temperature reaction systems because the material sintering at high temperature results in low specific surface area after the synthesis for all the materials prepared by solid-state method, which eliminates the sensitivity of results on the powder surface area. Then the differences in reaction kinetics are rather explained by the different redox properties of the materials than by the different morphology of the powders, particle size or porous structure.
Later on, thermochemical redox cycles were performed using these materials and the corresponding reduction and re-oxidation profiles obtained are shown in Fig. 1(a). Even though LCO exhibited high extent of reduction at 1300 °C (369 μmol g−1 of evolved O2), the extent of re-oxidation was lower as evidenced from the low CO yield during the first cycle (only 123 μmol g−1 of CO was produced during re-oxidation with CO2). This value got further reduced to 22 μmol g−1 during the second re-oxidation step indicating that both the activity and stability of LaCoO3 are significantly poor, because of extensive high-temperature sintering resulting in deactivation. The loss in reaction rate during oxidation is principally due to the diffusion limitations induced by the packing and densification of the particle layer inside the TG crucible, which creates an additional physical barrier to CO2 diffusion. This loss of activity always occurs mostly during the first cycle after powder packing in the crucible. Consequently, the first cycle is of major importance to highlight the deactivation process, which justifies focussing on the reactivity difference between the first and the second cycle.
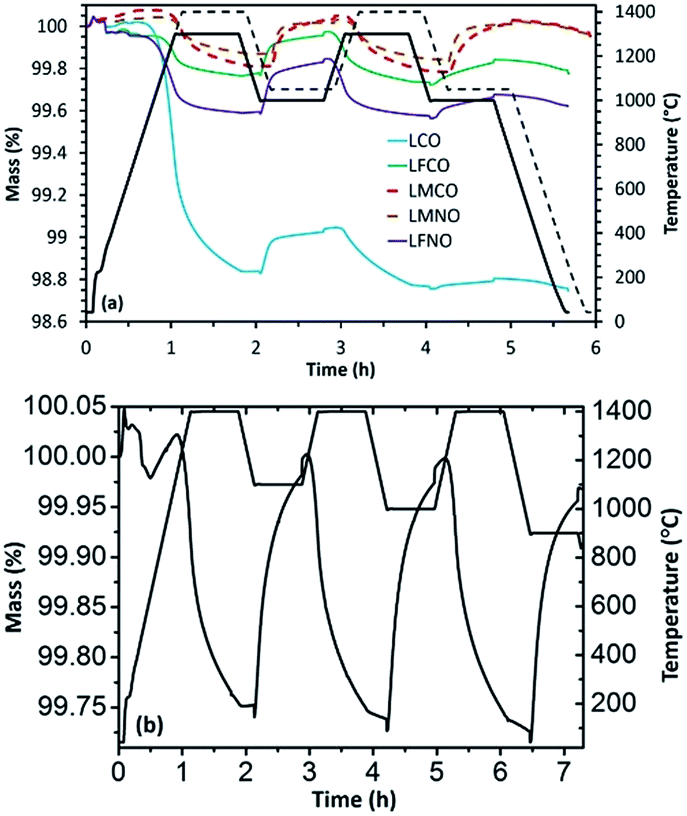 | ||
| Fig. 1 (a) Reduction and re-oxidation profiles as determined by performing TGA for the La(B,B′)O3 perovskites. (b) TGA of LaMn0.5Co0.5O3 with thermal reduction at 1400 °C in Ar and re-oxidation in 50% CO2/Ar at different oxidation temperatures (1100, 1000, and 900 °C). | ||
In the case of 50% Mn substituted material (LMCO), the extent of reduction (83 μmol g−1 of O2) was significantly lower than un-substituted LaCoO3. Interestingly, this value remained constant during the second cycle indicating total re-oxidation during the first cycle. The presence of Mn in the B-site considerably lowered the sintering issue, thereby enhancing the cycling performance stability. Moreover, the extent of re-oxidation was slightly enhanced and the amount of evolved O2 remained stable for the two cycles, as evidenced from Table 1. Also, LMCO displayed a remarkably stable redox activity for three cycles during which the re-oxidation temperatures were varied between 900 °C to 1100 °C, as evidenced from Fig. 1(b) (O2 production of 85, 82 and 85 μmol g−1 and CO production of 150, 154 and 154 μmol g−1, respectively). The re-oxidation rate remained unchanged while varying the temperature between 900 and 1100 °C suggesting the absence of kinetic limitations. The cyclability of this material was thus confirmed by the very similar re-oxidation profile with a CO production of the same magnitude (Fig. ESI-3†). The reduction and re-oxidation were carried out for a fixed duration of 45 min, and the fuel yield could even be enhanced with longer reaction durations as the equilibrium was not attained. Indeed, the mass loss or gain did not reach equilibrium, which means that further O2/CO could have been produced with longer reaction duration. However the goal was here to maintain reasonable cycling durations for practical implementation. It is thus not relevant using extended durations as this would not reflect realistic operating conditions used in real solar reactors. These durations were thus chosen to be able to perform cycles in a reasonable time and were typically high enough to reach significant reaction extent. The cycles were also performed to determine the reduction and oxidation capabilities at given reaction durations for a comparison purpose. Specifically, the reversibility of reactions was assessed to highlight to what degree the mass loss during reduction can be recovered during oxidation (oxidation yield).
| Material | T red (°C) | O2 evolved (μmol g−1) | CO evolved (μmol g−1) | ||
|---|---|---|---|---|---|
| 1st cycle | 2nd cycle | 1st cycle | 2nd cycle | ||
| a Temperature of reduction. | |||||
| LaCoO3 | 1300 | 369 | 86 | 123 | 22 |
| LaMn0.5Co0.5O3 | 1400 | 83 | 84 | 145 | 152 |
| LaFe0.75Co0.25O3 | 1300 | 59 | 74 | 117 | 65 |
| LaMn0.5Ni0.5O3 | 1400 | 54 | 59 | 97 | 112 |
| LaFe0.75Ni0.25O3 | 1300 | 114 | 84 | 150 | 62 |
The results further indicated that substitution with Fe in the B site (LFCO) altered the redox properties of the pure LaCoO3 since the amount of O2 produced (59 μmol g−1) during the first reduction step was significantly lower and CO (117 μmol g−1) evolved during the re-oxidation step remained similar. In the second cycle the amount of produced O2 increased slightly in comparison with the first cycle, however with a lower amount of CO produced because of material deactivation. Thus, Fe substitution did not contribute to improve the stability of LaCoO3 during thermochemical reduction and re-oxidation with CO2. The re-oxidation decrease was again mainly the result of diffusion limitations inside the packed bed of particles in the TG crucible, because of powder packing and densification induced by the thermal treatment. For LaMn0.5Ni0.5O3 and LaMn0.5Co0.5O3, the amounts of O2 and CO produced remained essentially constant during the two cycles of operation, unravelling the beneficial effect of Mn addition (Table 1). In contrast, LaFe0.75Ni0.25O3 (LFNO) did not exhibit stable thermochemical redox activity, similarly to LFCO.
The amounts of CO and O2 produced along with re-oxidation yields are compiled in Fig. 2, which compares the performances of the materials containing two metallic cations on the B site.
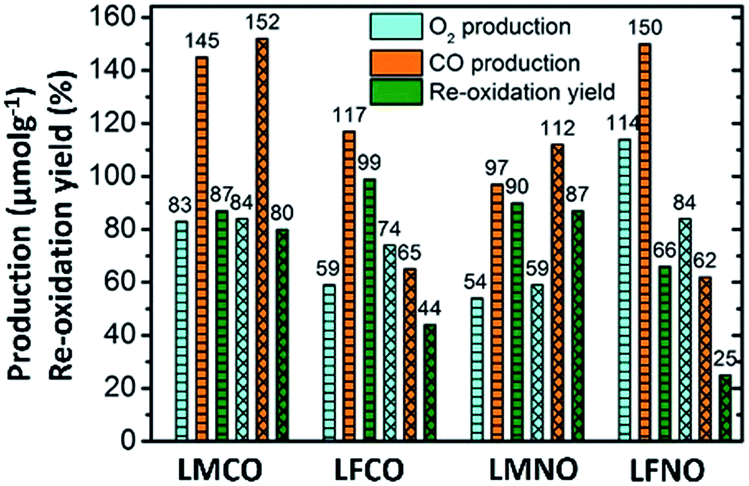 | ||
| Fig. 2 O2 and CO production along with re-oxidation yields for B-site substituted perovskites. | ||
Alkaline earth based perovskites, (A,A′)(B,B′)O3
Perovskite structured oxides with A-site occupied either partially or completely by alkaline earth cations constitute another interesting category of redox materials. In order to study the efficiency of alkaline earth based perovskites for solar thermochemical fuel production, Ba and Sr containing perovskite oxides were synthesized by the solid-state method. SrFeO3 (SFO) materials were obtained after performing calcination at 1300 °C during synthesis. In order to analyse the effect of barium, Ba0.5Sr0.5FeO3 (BSFO) was synthesized at a calcination temperature of 1100 °C. The composition of this material was further modified by incorporating 80% Co in the B site of the perovskite structure forming Ba0.5Sr0.5Fe0.2Co0.8O3 (BSFCO). In this case, a calcination temperature of 1000 °C was used for perovskite phase formation. The synthesis of materials with perovskite structure was confirmed from XRD (Fig. ESI-4†). Thermochemical redox cycles were then performed using these materials and the obtained results are summarized in Table 2 and Fig. 3. Materials such as SrFeO3 and Ba0.5Sr0.5FeO3 are known to have mixed Fe valence with the presence of Fe4+. Due to the elevated initial oxidation state of these metallic cations, which is superior to those observed in stable Fe-containing binary oxides, these perovskites represent an interesting category of non-stoichiometric oxides to investigate, which start releasing large extents of O2 at lower temperatures than most compounds considered in the field of two-step thermochemical splitting of CO2, thereby possibly lowering the temperatures necessary to achieve CO production.| Material | T red (°C) | O2 evolved (μmol g−1) | CO evolved (μmol g−1) | ||
|---|---|---|---|---|---|
| 1st cycle | 2nd cycle | 1st cycle | 2nd cycle | ||
| a Temperature of reduction. | |||||
| SrFeO3 (SFO) | 1200 | 795 | 47 | 100 | 72 |
| SrFeO3 (SFO-1) | 1100 | 782 | 52 | 96 | 69 |
| Ba0.5Sr0.5FeO3 | 1000 | 582 | 31 | 136 | 78 |
| Ba0.5Sr0.5Fe0.2Co0.8O3 | 1000 | 550 | — | — | — |
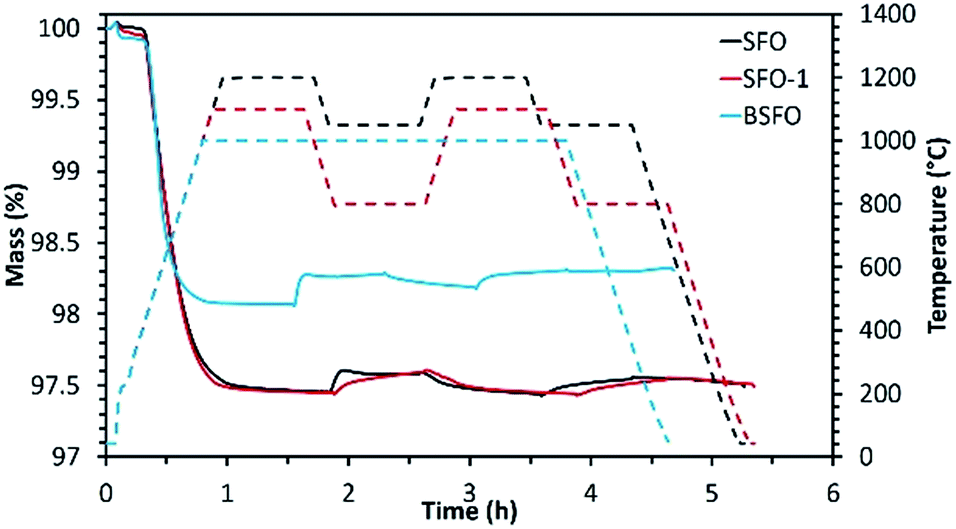 | ||
| Fig. 3 Reduction and re-oxidation profiles as determined by performing TGA for the alkaline earth based perovskites. | ||
Initial studies in this series were performed using SFO. Reduction was performed at 1200 °C followed by re-oxidation at 1050 °C. Even though in this case, extremely high extent of reduction (795 μmol g−1 of O2) was achieved during the first cycle, the extent of re-oxidation was significantly lower as evidenced from the low CO yield. In the first cycle only 100 μmol g−1 of CO was produced during re-oxidation of SFO with CO2. This value got further reduced to 72 μmol g−1 during the second re-oxidation step, indicating poor performance of this material. During another test performed using the same material (SFO-1), the reduction and re-oxidation temperatures were decreased to 1100 °C and 800 °C respectively. In this case also (SFO-1), in spite of the initial high extent of reduction (782 μmol g−1 of O2), CO yields remained rather low (96 μmol g−1 and 69 μmol g−1 for the first and second re-oxidation steps, respectively) and similar to the previous test as mentioned above. These results prove that the redox performance of SrFeO3 is not significantly dependent of the reduction and re-oxidation temperatures. Indeed, low extent of sintering was observed for this composition in both tests. For BSFO, reduction and re-oxidation were performed under isothermal conditions (1000 °C). Partial substitution with 50% Ba diminished the initial extent of reduction producing 582 μmol g−1 of O2. Even though CO produced during the first re-oxidation step increased to 136 μmol g−1, this value got decreased to 78 μmol g−1 during the second cycle. Interestingly carbonate formation typical of Ba containing perovskites, was not observed in this case (Fig. ESI-5(a)†). Further studies were also performed to monitor the effect of Co substitution in Ba0.5Sr0.5FeO3 by incorporating 80% Co in the structure denoted as BSFCO. Extremely low CO production (<10 μmol g−1) was observed under isothermal conditions of reduction and re-oxidation at 1000 °C for this material. This can be attributed to sintering and favoured carbonate formation under the reaction conditions (Fig. ESI-5(b)†), as evidenced by the large mass gain observed during exposure to CO2 (Fig. ESI-6†).
For this series, it can be summarized that these materials exhibit a high reducibility, enabling the decrease of the thermochemical cycling temperatures by several hundred degrees while maintaining performances (CO yields) similar to those of CeO2. However, these perovskites generally show a lower thermal stability. BSFCO is not suitable as it decomposes under CO2 at high temperature to form carbonate species, as confirmed by the XRD analysis of the recovered material after cycling (Fig. ESI-5(b)†). The most chemically stable compounds (generally containing more Fe) show important sintering issues causing material deactivation during cycling. Thus, despite promising performances during first cycle, optimization of the cycling stability from microstructure tuning will be required for using such materials in solar thermochemical applications.
Rare earth – alkaline earth based perovskites (La,Sr)(B,B′)O3
A combination of rare-earth and alkaline-earth cations was included in the A-site of the perovskite structure to study its effect on the efficiency and stability with respect to sintering and carbonate formation. La0.6Sr0.4FeO3 (LSFO), La0.5Sr0.5MnO3 (LSMO), La0.5Ca0.5MnO3 (LCMO), Y0.5Sr0.5MnO3 (YSMO), La0.6Sr0.4Co0.2Fe0.8O3 (LSCFO), La0.5Sr0.5Mn0.5Fe0.5O3 (LSMFO) and La0.5Sr0.5Mn0.5Co0.5O3 (LSMCO) were synthesized by solid-state method at calcination temperatures of 1300 °C (or 1400 °C for Mn containing perovskites). In each case, the formation of perovskite phase was confirmed from the wide angle powder XRD patterns given in Fig. ESI-7.†The results obtained from the thermochemical redox cycling experiments are compiled in Fig. 4 and Table 3. In the case of La0.6Sr0.4FeO3, reduction was carried out at 1200 °C and re-oxidation at 1050 °C. Although high reduction extent was achieved because of the presence of reducible mixed valence Fe3+/Fe4+ cations, the amount of CO produced was low and decreased from 53 μmol g−1 to 18 μmol g−1 during the second cycle indicating that this material is not stable. Such low amount of gas production can be attributed to the high degree of sintering under the reaction conditions. Even though significant amount of CO evolution was observed for La0.5Sr0.5MnO3 and La0.5Ca0.5MnO3, the amount got decreased indicating stability issues. In this case, reduction and re-oxidation cycles were performed at 1400 °C and 1050 °C, respectively. For the same operating conditions, Y0.5Sr0.5MnO3 showed high reduction extent but low re-oxidation yield (∼10%), although the overall CO production (above 100 μmol g−1) was still comparatively higher than the one obtained for CeO2 (maximum CO production from ceria is ∼100 μmol g−1 after thermal reduction at 1400 °C in Ar under the same oxidizing conditions, and since ceria is totally re-oxidized this fuel productivity cannot be improved unless increasing the reduction temperature for enhanced oxygen vacancies formation). This favourable comparison with a well-known reference material gives insights into the performances of the considered perovskites (fuel production capabilities) using the same experimental method and configuration. SEM analysis indicated significant sintering for the synthesized material compositions without any strong modification of the structure after the redox cycling experiments (Fig. ESI-2†). La0.6Sr0.4Co0.2Fe0.8O3 was analysed under a reduction temperature of 1200 °C and a re-oxidation temperature of either 800 °C or 1050 °C under CO2. In this case initial reduction was slightly improved (in comparison with La0.6Sr0.4FeO3) producing 465 μmol g−1 of O2. However, the CO produced during the re-oxidation step still remained low regardless of the oxidation temperature (62 and 78 μmol g−1 at 800 and 1050 °C respectively) and further decreased markedly during the second cycle. Hence LSFO and LSCFO materials exhibit low performance capabilities for CO2-splitting.
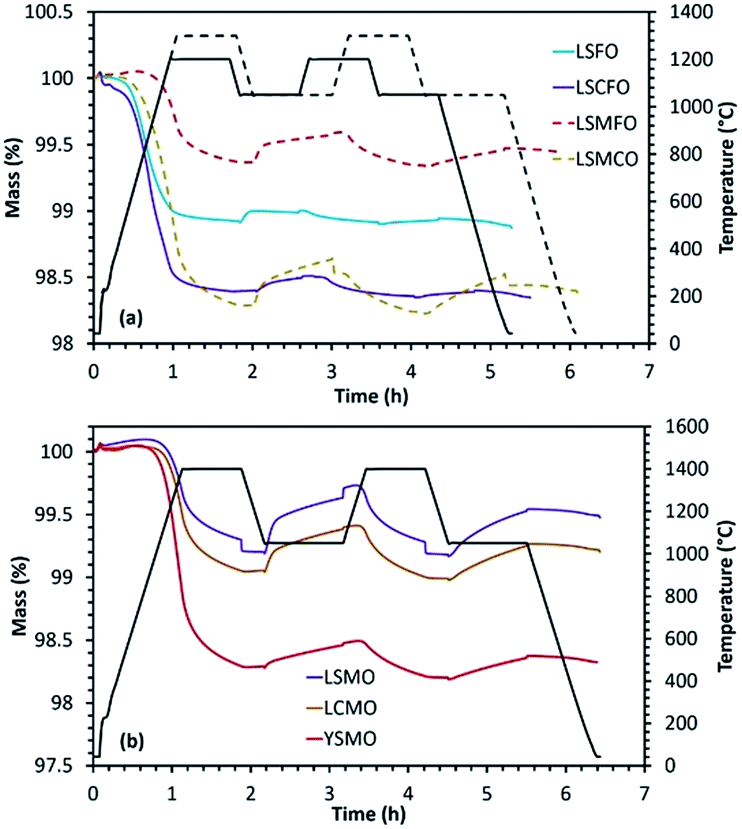 | ||
| Fig. 4 (a) Reduction and re-oxidation profiles as determined by performing TGA for (La,Sr)(B,B′)O3 perovskites and (b) TGA of La0.5Sr0.5MnO3, La0.5Ca0.5MnO3 and Y0.5Sr0.5MnO3 with thermal reduction at 1400 °C and re-oxidation at 1050 °C. | ||
| Material | T red (°C) | O2 evolved (μmol g−1) | CO evolved (μmol g−1) | ||
|---|---|---|---|---|---|
| 1st cycle | 2nd cycle | 1st cycle | 2nd cycle | ||
| a Temperature of reduction. | |||||
| La0.6Sr0.4FeO3 | 1200 | 337 | 27 | 53 | 18 |
| La0.5Sr0.5MnO3 | 1400 | 248 | 141 | 269 | 215 |
| La0.5Ca0.5MnO3 | 1400 | 311 | 132 | 210 | 168 |
| Y0.5Sr0.5MnO3 | 1400 | 551 | 90 | 112 | 105 |
| La0.6Sr0.4Co0.2Fe0.8O3 | 1200 | 465 | 46 | 62 | 24 |
| La0.6Sr0.4Co0.2Fe0.8O3 | 1200 | 454 | 47 | 78 | 29 |
| La0.5Sr0.5Mn0.5Fe0.5O3 | 1300 | 214 | 78 | 135 | 79 |
| La0.5Sr0.5Mn0.5Co0.5O3 | 1300 | 538 | 91 | 152 | 125 |
A combination of Mn and Fe cations in the B site (La0.5Sr0.5Mn0.5Fe0.5O3) was later analysed under the reduction and re-oxidation temperatures of 1300 °C and 1050 °C respectively. The CO yield increased to 135 μmol g−1 during the first cycle which then decreased to 79 μmol g−1 during the second cycle indicating low stability. However the CO produced during the second cycle was still higher compared to other members in the series. Finally, the performance of La0.5Sr0.5Mn0.5Co0.5O3 was monitored under reduction and re-oxidation temperatures of 1300 °C and 1050 °C, respectively. The amount of produced CO significantly increased to 152 μmol g−1 and 125 μmol g−1 during the first and second cycles. However, the decrease of CO amount was the result of sintering and carbonate formation was noticed during exposure to CO2 (about 30% of the mass gain can be attributed to carbonate formation). The carbonate species are then decomposed swiftly when stopping CO2 injection and switching to the inert gas for the subsequent reduction step (Fig. 4(a)), case of LSMCO. The aforementioned studies clearly indicate higher CO production for LSMO, however with lack of stability. Therefore with the objective of improving the stability while maintaining the performance, the effect of La and Sr in the structure was monitored by individually replacing these cations with Y and Ca, respectively (Fig. 4(b)). LCMO displayed better reducibility, but the amount of evolved CO was lower in comparison with LSMO. Correspondingly, YSMO exhibited high reducibility but low CO evolution. Similar observations were previously made for these three compositions synthesized by the Pechini method. It is interesting to note that, the materials synthesized by the Pechini method with a porous microstructure were found to be more stable in comparison with the ones synthesized by the solid-state method.25
It is generally admitted that the CO production capability depends largely on the materials ability to release oxygen, and that the reduction yield and the O2 production rate determine the materials performance. This study shows that it is not always true for most of the perovskite materials (see for example YSMO that stands out by large reduction capabilities but low re-oxidation yields). Since the re-oxidation is only partial because of kinetic limitations and of a thermodynamic driving force that becomes lower as δ decreases when oxygen is replenished in the oxide lattice, it is then perfectly normal that the amount of CO produced is not twice the amount of O2 produced during the reduction.12 Longer reaction durations would somewhat result in the increase of the CO to O2 ratio but to a small extent only. This is consistent with the fact that the materials that reduce readily will commonly be hardly oxidized with CO2 because of thermodynamic limitation, whereas awkward reduction abilities will generally result in prompt oxidation, ceria being a prototypal example of this latter behaviour. Then, any studies reporting both high reduction and outstanding re-oxidation capabilities should be considered cautiously regarding their data reliability. The reduction is a thermally activated process that proceeds with continuous topotactic oxygen disincorporation from the oxide lattice. The thermodynamic driving force is decreased during the oxidation process as the oxygen vacancies are refilled and the non-stoichiometry is decreased. The thermodynamic barrier can be counteracted by using large oxidant excess to shift the reaction equilibrium favouring fuel production or by large temperature swing to activate the re-oxidation step at the expense of kinetic limitations that may arise and additional heat losses (these solutions are thus not acceptable for a solar process).
Regarding the benefits of Sr-substituted perovskites (e.g.: LSMO), the reduction extent increases with Sr2+ substitution for La3+. Based on charge balance considerations, the increase of Sr2+ content on the A-site results in a higher nominal oxidation state of the cation on the B-site of the perovskite. The higher the Mn4+ content, the more favoured the reduction of the compounds, which results in the tendency observed here within the LSMO series (Fig. 5). Previous studies suggest that the oxidation thermodynamics (ΔGox) should be more favourable for CeO2, especially when the Sr content is increased in LSMO compounds.12 Therefore, the significant CO productions observed here mainly result from the strong reduction capability of the perovskite structure, allowing significant CO evolution despite incomplete re-oxidation yields (for the first cycle: 54% for LSMO versus complete re-oxidation yield for CeO2 at the same temperature, reaction duration and oxidant molar flow rate). The evolution of the mean Mn oxidation state can be calculated by charge balance from the weight variations observed during the cyclic experiments (Fig. ESI-8†), since Mn is the only redox active cation in the La1−xSrxMnO3−δ. In accordance with the presence of mixed valence Mn4+/Mn3+, thermal redox cycle should involve the change of Mn4+ into Mn3+, and vice versa. Fig. 5(b) shows the evolution of Mn oxidation state in LSMO series during the first thermochemical cycle, based on the initial stoichiometry for the as made materials. At the start of the cycle, Mn oxidation state is ranging from +3.35 to +3.80 as x is increased from 35 to 80%, before being decreased during thermal reduction and increased during the CO2-splitting step. Indeed, final reduced oxidation states of Mn are +3.29 (LSMO35), +3.31 (LSMO50), +3.31 (LSMO65), and +3.26 (LSMO80), while the initial oxidation states are +3.35, +3.50, +3.65, and +3.80 respectively. These final stoichiometries denote a very narrow range of reduced Mn oxidation states, regardless of the Sr content and associated oxygen release. This similarity is also observed during the CO2 dissociation step, which also results in a very narrow range of final oxidation states after the re-oxidation of the LSMO compounds, with extreme values of +3.33 for LSMO80 and +3.42 for LSMO50. Such comparable redox activity of Mn for all compounds, explains why similar CO productions are achieved within the series despite a strong discrepancy in the re-oxidation yields.
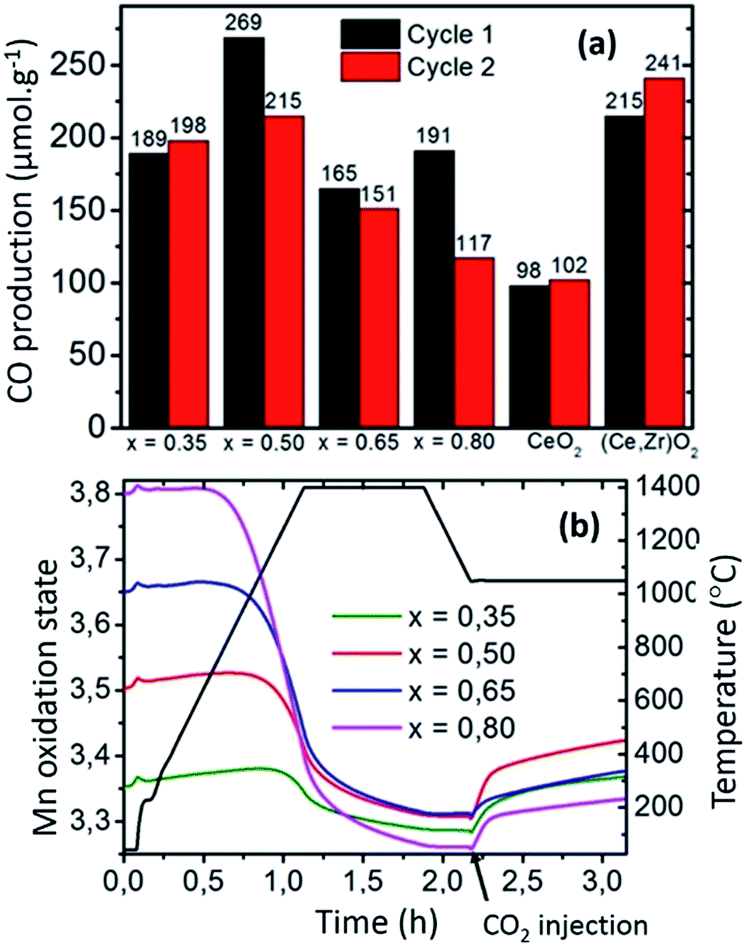 | ||
| Fig. 5 (a) CO productions during two cycles for La1−xSrxMnO3 series and comparison with fluorite materials as reference (oxidation at 1050 °C after thermal reduction at 1400 °C in Ar). (b) Evolution of the valence state of manganese during the thermal reduction in Ar and re-oxidation with CO2. | ||
For example, the similar Mn oxidation states reached after both the reduction and oxidation for LSMO35 and LSMO80 allows them to produce respectively 189 and 191 μmol g−1, whereas the associated re-oxidation yields are 92 and 14%. Thus, the final CO production capability is not directly governed by the final oxygen non-stoichiometry achieved depending on the distinct oxygen vacancy formation capabilities of the compounds, but it is rather governed by the final oxidation state of the redox-active Mn cation obtained after reduction.
Ruddlesden–Popper mixed oxides
Another category of mixed oxides with interesting redox properties belongs to the Ruddlesden–Popper structure. However, such materials are the least explored for solar thermochemical fuel production so far. We performed here a comparison of the results obtained for perovskites with a series of Ruddlesden–Popper structured materials. For this purpose, LaSrCoO4 (LSCO-RP1), LaSrCo0.67Fe0.33O4 (LSCFO-RP1), LaSrCo0.67Mn0.33O4 (LSCMO-RP1), La0.5Sr1.5MnO4 (LSMO-RP1) and LaSr2Mn2O7 (LSMO-RP2) were studied. These materials were calcined at 1400 °C, except LaSrCo0.67Mn0.33O4 that was synthesized using a calcination temperature of 1500 °C. For all these materials, crystalline peaks in accordance with the corresponding composition were observed in the wide angle region of the XRD patterns provided in Fig. ESI-9 and 10.† The results obtained from thermochemical reduction and re-oxidation profiles are given in Fig. 6 and Table 4. LaSrCoO4 was analysed under a reduction temperature of 1300 °C and a re-oxidation temperature of 1050 °C. High sintering and carbonate formation was observed (about 61% of the mass gain during exposure to CO2). The carbonate species were then decomposed promptly when switching to the inert gas during the second reduction step. By incorporating Fe in the B-site (LSCFO-RP1), high sintering was still observed, and carbonate formation was again evidenced (about 49% of the mass gain was ascribed to carbonate formation). Likewise, Mn incorporation in place of Fe (LSCMO-RP1) resulted in similar behaviour with extremely high sintering (approaching melting) and carbonate formation, under the same conditions of reduction and re-oxidation. RP phases with single Mn alone on the B site were then investigated (Fig. 6(a)). In the case of La0.5Sr1.5MnO4 (RP1 phase), noticeable performance stability was observed with about 172 μmol g−1 of CO produced during both the first and second re-oxidation steps, indicating high extent of stability for this material and absence of carbonate formation. Likewise, LaSr2Mn2O7 (RP2 phase) produced 206 μmol g−1 and 191 μmol g−1 of CO during the first and the second cycle, thus exhibiting noticeable performance stability and absence of carbonate formation. The re-oxidation yield was also improved (75% for La0.5Sr1.5MnO4 and 60% for LaSr2Mn2O7 in the first cycle) when compared with La0.5Sr0.5MnO3 (54%) (Fig. ESI-11†). In addition, these Mn-based RP phases did not show any evidence of sintering, consistently with the observed redox stability. Finally, the results obtained with parent RP phases show similar characteristics as those of LSMO perovskites containing the same elements.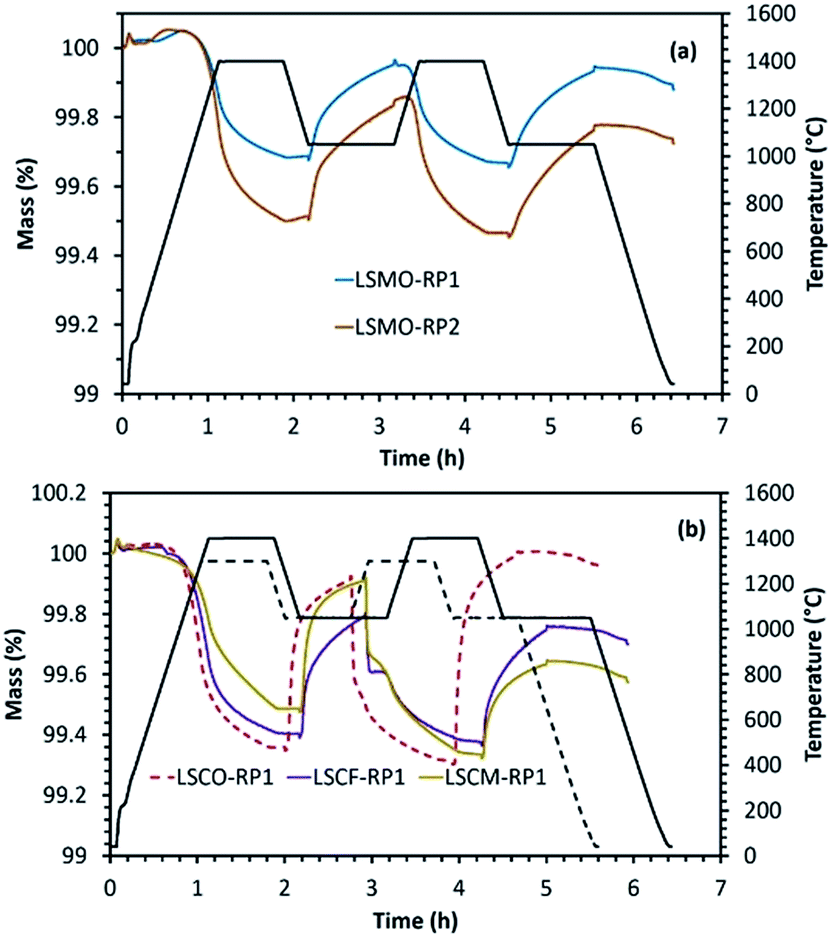 | ||
| Fig. 6 (a) Reduction and re-oxidation profiles as determined by performing TGA for Ruddlesden–Popper materials. (b) TGA of Co-containing RP materials with carbonate formation occurring during the exposure to CO2. | ||
| Material | T red (°C) | O2 evolved (μmol g−1) | CO evolved (μmol g−1) | ||
|---|---|---|---|---|---|
| 1st cycle | 2nd cycle | 1st cycle | 2nd cycle | ||
| a Temperature of reduction. | |||||
| LaSrCoO4 | 1300 | 210 | 84 | 139 | — |
| LaSrCo0.67Fe0.33O4 | 1400 | 187 | 72 | 128 | — |
| LaSrCo0.67Mn0.33O4 | 1400 | 160 | 103 | 111 | — |
| La0.5Sr1.5MnO4 | 1400 | 115 | 88 | 172 | 173 |
| LaSr2Mn2O7 | 1400 | 173 | 123 | 206 | 191 |
This class of materials thus appears to be promising with regard to the flexibility of the phases, ability to accommodate non-stoichiometry, and stabilization of mixed valences; thereby allowing redox properties similar to those of perovskites. RP phases are also subjected to significant topotactic oxygen disincorporation due to a crystal chemistry that is reminiscent of that of the perovskite. They are frequently referred to layered perovskites, owing to their parent crystal structure. For the RP phases, lower O2 productions were observed when compared to LSFO and LSCFO reduced at 1200 °C, which is partly related to an initial oxidation state of +3 for B in the LaSrBO4 phases, while the oxidation state was greater in LSFO, LSCFO, LSMO and LSMCO. The presence of Co generally favours a larger weight loss when comparing LSCFO to LSFO, in agreement with cobalt oxides generally reducing more easily than iron oxides for an equal oxidation state. Regarding the RP phases, the largest O2 production is observed during the reduction of LSCO-RP1. Similarly to the comparison of LSCFO versus LSFO, the cobalt containing LaSrCoO4 shows greater reducibility at high temperature than the iron containing one. However cobalt containing RP phases showed intermediate carbonate formation during exposure to CO2.
In order to evidence the existence of carbonate formation, a redox cycle was performed with LaSrCoO4 subjected to thermal reduction in Ar at 1300 °C and oxidation in 20% O2/Ar at 1050 °C (Fig. 7). Replacing CO2 by O2 during the oxidation step allows eliminating the carbonate formation. As a result, it can be observed that the reduction profile in the subsequent cycles differs substantially from the one observed when using CO2 as oxidant. In this case, the reduction shows a smoothed oxygen evolution (mass loss) profile very similar to the one of the first reduction. In contrast, when using CO2 as oxidant inducing carbonate formation as side product, the subsequent reduction step is much different from the initial one. In this case, the next reduction step following CO2 oxidation step commonly features a steep mass decrease attributed to rapid decarbonation followed by a slower mass decrease attributed to thermal reduction. This is valid whenever carbonate formation occurs regardless of the material (case of LSMCO, LSCO-RP1, LSCFO-RP1, LSCMO-RP1). This adverse side reaction lowers the materials activity and the fuel productivity, which is thus unsuitable for efficient thermochemical CO2-splitting.
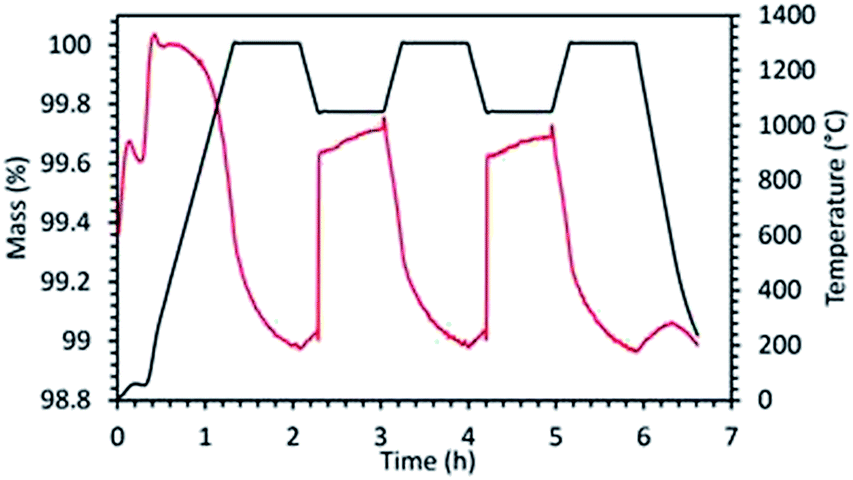 | ||
| Fig. 7 TGA of LaSrCoO4 during reduction in Ar at 1300 °C and re-oxidation in 20% O2/Ar at 1050 °C. | ||
Discussion
Manganite perovskites appear suitable for favouring both thermal and chemical material stability. In LSMO perovskites, the variation of Mn oxidation state implies a partial oxidation of Mn3+ into Mn4+ during the CO2-splitting step. The same redox activity can be observed in the parent RP phases (La0.5Sr1.5MnO4, LaSr2Mn2O7) with initial Mn oxidation state of +3.50. Similarly, starting at +3 for Co valence in LaSrCoO4, cobalt oxidation state variation in the RP phase during redox process means that the Co3+/Co2+ redox couple is active. Therefore, perovskite and RP materials enable the activation for CO2-splitting of redox couples that are not available in stoichiometric oxides and that commonly show highly unfavourable thermodynamics. These materials thus offered noticeable access to metallic valence transitions during re-oxidation in CO2 atmosphere that are fundamentally not active in stoichiometric oxides. They also display significant ionic conductivity at high temperature that is beneficial for bulk diffusion processes involved during both the thermal reduction and the CO2-splitting steps.Table 5 gathers the maximum rates of O2 and CO production for each material as well as the temperatures of maximal O2 production (peak rate of O2 production). These rates were obtained from the derivative of the mass variation. The O2 production rate shows a peak during the heating period whatever the sample composition, which corresponds to the maximum rate of thermal reduction (the temperatures of peak reduction rate are reported in Table 5). In contrast, the CO production rate is always maximal as soon as CO2 is injected and it then gradually decreases. Thus the maximal rates of CO production reported in Table 5 correspond to the initial rates of the oxidation reaction at the time of CO2 injection.
| Material | T peak-red (°C) | Peak O2 production rate (μmol g−1 s−1) | Peak CO production rate (μmol g−1 s−1) | ||
|---|---|---|---|---|---|
| 1st cycle | 2nd cycle | 1st cycle | 2nd cycle | ||
| a Temperature of maximal rate of thermal reduction (peak rate of O2 production). | |||||
| LaCoO3 | 1292 | 0.359 | 0.063 | 0.288 | 0.034 |
| LaMn0.5Co0.5O3 | 1397 | 0.093 | 0.098 | 0.187 | 0.178 |
| LaFe0.75Co0.25O3 | 1292 | 0.073 | 0.082 | 0.276 | 0.069 |
| LaMn0.5Ni0.5O3 | 1397 | 0.066 | 0.066 | 0.299 | 0.282 |
| LaFe0.75Ni0.25O3 | 1285 | 0.102 | 0.098 | 0.306 | 0.110 |
| SrFeO3 (SFO) | 492 | 0.734 | 0.057 | 0.419 | 0.070 |
| SrFeO3 (SFO-1) | 477 | 0.731 | 0.056 | 0.162 | 0.068 |
| Ba0.5Sr0.5FeO3 | 549 | 0.975 | 0.079 | 0.953 | 0.282 |
| Ba0.5Sr0.5Fe0.2Co0.8O3 | 460 | 0.724 | — | — | — |
| La0.6Sr0.4FeO3 | 807 | 0.229 | 0.034 | 0.169 | 0.034 |
| La0.5Sr0.5MnO3 | 1398 | 0.230 | 0.172 | 0.459 | 0.159 |
| La0.5Ca0.5MnO3 | 1386 | 0.289 | 0.127 | 0.316 | 0.096 |
| Y0.5Sr0.5MnO3 | 1367 | 0.416 | 0.064 | 0.163 | 0.052 |
| La0.6Sr0.4Co0.2Fe0.8O3 | 844 | 0.256 | 0.049 | 0.161 | 0.036 |
| La0.5Sr0.5Mn0.5Fe0.5O3 | 1290 | 0.199 | 0.079 | 0.265 | 0.050 |
| La0.5Sr0.5Mn0.5Co0.5O3 | 1273 | 0.318 | 0.074 | 0.314 | 0.087 |
| LaSrCoO4 | 1290 | 0.189 | — | — | — |
| LaSrCo0.67Fe0.33O4 | 1391 | 0.155 | — | — | — |
| LaSrCo0.67Mn0.33O4 | 1398 | 0.116 | — | — | — |
| La0.5Sr1.5MnO4 | 1398 | 0.108 | 0.100 | 0.199 | 0.147 |
| LaSr2Mn2O7 | 1391 | 0.176 | 0.150 | 0.283 | 0.137 |
The gas yield data (O2 and CO production capacity) are the most relevant performance indicators for materials since the ultimate objective is the fuel production ability per unit mass of active material. However, the rate of gas production is also important but may be misleading when reported alone because O2/CO production rates and fuel productivities are not correlated. A material may indeed exhibit high initial CO production rate but low overall fuel productivity (case of ceria or BSFO for example).
Fig. 8 shows the evolution of the O2 production rate during the first thermal reduction step for La(B,B′)O3. The maximal O2 production rate is observed for LaCoO3. In contrast, the Mn-containing materials (LMCO, LMNO) show a peak rate of O2 production occurring at higher temperature (near 1400 °C), and their O2 and CO production rates are noticeably stable during cycling (Table 5). Besides, the materials with the highest reduction ability (namely, SFO, BSFO, BSFCO, LSFO, LSCFO) exhibit a peak rate of O2 production at the lowest temperatures (TBSFCO < TSFO < TBSFO < TLSFO < TLSCFO, as illustrated in Fig. ESI-12†). Regarding LSMO and related RP phases (LSMO-RP1, LSMO-RP2), the maximal rate of O2 production occurs at the same temperature and the decrease in the O2 release is only initiated once the samples reach the temperature plateau at 1400 °C (Fig. ESI-13†). This means that higher O2 production rates could be reached at higher reduction temperatures and that the reduction is controlled by the heating rate.
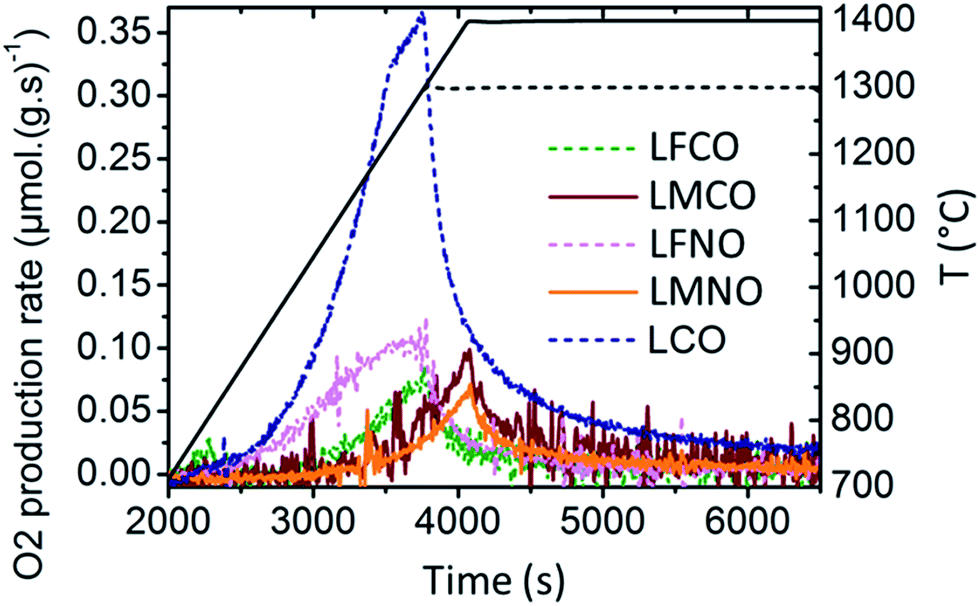 | ||
| Fig. 8 O2 production rate during the first thermal reduction step for La(B,B′)O3 materials. | ||
The reduction step is generally composed of a fast initial step assigned to surface reduction followed by a slower reduction rate during temperature plateau most corresponding to bulk oxygen release. Its duration is thus controlled by the heating rate that is fixed by the TG apparatus (here 20 °C min−1). The re-oxidation is always more sensitive to sintering than the reduction because of increased mass transfer limitations in the powder layer. The sintering chiefly induces mass transfer limitation in the powder bed. This is because the powder at the surface of the crucible reacts faster than the bottom of the crucible because of the TG configuration. The oxidation step is thus affected by inter-granular gas diffusion limitation in the material layer during oxidation with CO2 (external mass transfer limitation in the crucible), whereas oxygen ionic diffusion in the bulk material is not the rate limiting mechanism. These diffusion limitations could be alleviated by suitable materials shaping (for increasing the solid–gas surface area), thus reducing the necessary duration of the oxidation duration in a solar process. The reaction durations are thus highly dependent on the experimental system configuration and the reactant morphology.
The reduction extent provides information on the reduction capability when comparing the compounds relatively to one another (at T and pO2 fixed). The results indicate that the yield of thermal reduction is generally not altered by the sintering issue since the mass loss in the second cycle is generally higher or at least equal to that measured in the first cycle, which thus confirms that bulk solid-state diffusion is not limiting in the redox process. This provides compelling evidence about the existence of external mass transfer limitation only during the oxidation step (inter-granular CO2 diffusion). So the limitation for the CO production step is not intrinsic to the material but it is rather related to the system configuration.
It must be noted that the reactions were performed in TG using a packed bed of particles. The particles confined within the Pt-crucible naturally sit in small pile. This pile did not exceed 2–3 mm height in the experiments to limit the diffusion effects in the solid pile. However, the packing of the powder inside the crucible after the first thermal cycle enhanced the diffusion effects. As a result, the kinetic rate of the second oxidation may be lowered for some materials due to diffusion limitations inside the powder. This oxidation reaction was thus not finished and further CO could have been produced if the reaction duration was longer. The TG data usually show that the sample mass initially increased promptly during the oxidation, which denotes a reaction-controlled regime in the first few minutes when the materials regained the main part of their lost mass in the previous reduction step. However, the reduction reaction performed at higher temperatures (e.g. 1400 °C) induced powder packing and sintering in the crucible. As a result of these physical limitations, the sample mass uptake during the oxidation reaction was composed of a fast initial step as soon as CO2 was injected, followed by a diffusion-controlled step and CO production was still continuing after 45 min of reaction.
The cycling time was selected so that to provide suitable trade-off for reaching significant reaction extent while maintaining reasonable reaction duration for practical implementation in e.g. solar reactors. The cycle duration was not optimized in TG since the powder was reacting as a packed bed and the mass change did not reach steady state in some cases mainly because of diffusion limitations. It is likely that the reaction duration can be reduced with appropriate materials shaping (e.g. using porous ceramic foam or porous structures) for enhancing the solid–gas contact and exchange area. The thermochemical performance data shown here were collected at various temperatures taking into account the thermal stability and the melting point of the compounds. On the basis of annealing tests performed before redox cycles at various temperatures, it was observed for instance that LSMO compounds had a higher melting point than BSFO compounds. This is consistent with the fact that the synthesis of LSMO structure via solid-state reaction is commonly performed at 1400 °C or above, whereas very well crystallized BSFO structure can be obtained at 1000 °C using the same synthesis technique. This discrepancy in melting points strongly conditioned the choice for reduction and CO2-splitting temperatures. On the one hand, reduction temperatures were selected to achieve a significant reduction extent while remaining below the melting point for each formulation. This trade-off is necessary for obtaining maximal reduction potential while avoiding extensive material sintering that would alter the reactivity during solid–gas re-oxidation step. This explains why materials such as BSFCO are reduced at 1000 °C while others such as LSMO are reduced at 1400 °C. On the other hand, CO2-splitting temperatures were selected below the reduction temperatures to provide a favourable thermodynamic potential. Because of kinetic limitations, the oxidation temperature must be kept high enough, which concomitantly promotes oxide ion bulk diffusion (a key requirement for the redox activity of non-stoichiometric compounds). These temperatures thus resulted from a trade-off and were not definitely optimized. The continuous oxygen evolution with temperature encountered for perovskites differs from stoichiometric oxides for which the reduction proceeds via discrete phase transitions with well-defined oxygen evolution. Therefore, optimization of the thermal cycling conditions for the most efficient perovskites should be conducted to further enhance their CO2-splitting capabilities. It can be also expected that noticeable improvements in the performance of these materials may be achieved via the tuning of their morphology and microstructure (linked to the synthesis route), chemical composition (content of each element in the perovskite) and temperatures applied to the thermochemical cycles.
Conclusions
The solar thermochemical two-step dissociation of CO2 based on perovskite compounds was investigated to identify the most suitable materials compositions for this application. A comprehensive study was performed to understand the variations in the redox activity of perovskites and related Ruddlesden–Popper (RP) structured oxides, with respect to the variations in composition and temperature. The investigated materials showed significant CO productions, especially La(B,B′)O3 perovskites with (Mn, Ni) or (Mn, Co) together in the B-site, as well as Sr-doped lanthanum manganites with perovskite and parent RP structures, comparing favourably to ceria. For La-based B-site substituted perovskites (La(B,B′)O3), the presence of Mn in the B-site lowered the extent of sintering, with a constant re-oxidation rate between 900 and 1100 °C. The best performance was observed for LaMn0.5Co0.5O3 with CO productions over 150 μmol g−1 per cycle. Regarding alkaline earth based perovskites, (A,A′)(B,B′)O3, carbonate formation was observed in the case of Ba0.5Sr0.5Fe0.2Co0.8O3. For other compositions such as Ba0.5Sr0.5FeO3 and SrFeO3, low re-oxidation extent was achieved. Mn-containing perovskites (LaxSr1−xMnO3) showed the highest CO production capabilities, which was governed by the final oxidation state of the redox-active Mn cation obtained after reduction. Likewise, Ruddlesden–Popper materials with only Mn in the B-site (La0.5Sr1.5MnO4, LaSr2Mn2O7), exhibited noteworthy CO production capacity (over 200 μmol g−1) and stability during thermochemical two-step redox cycling, without any microstructural or morphological optimization. Such new formulations thus constitute promising class of redox materials in the context of solar thermochemical fuel production and CO2 valorisation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the French National Agency for Research (ANR, SUNFUEL project, contract no. ANR-16-CE06-0010-01).Notes and references
- C. Perkins, P. R. Lichty and A. W. Weimer, Int. J. Hydrogen Energy, 2008, 33, 499–510 CrossRef CAS.
- A. Steinfeld, Int. J. Hydrogen Energy, 2002, 27, 611–619 CrossRef CAS.
- M. D. Allendorf, Energy Fuels, 2008, 22, 4115–4124 CrossRef CAS.
- A. Steinfeld, Sol. Energy, 2005, 78, 603–615 CrossRef CAS.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797–1801 CrossRef CAS PubMed.
- P. Furler, J. R. Scheffe and A. Steinfeld, Energy Environ. Sci., 2012, 5, 6098–6103 CAS.
- S. Abanades, A. Le Gal, A. Cordier, G. Peraudeau, G. Flamant and A. Julbe, J. Mater. Sci., 2010, 45, 4163–4173 CrossRef CAS.
- B. Bulfin, F. Call, J. Vieten, M. Roeb, C. Sattler and I. V. Shvets, J. Phys. Chem. C, 2016, 120, 2027–2035 CAS.
- A. Le Gal, S. Abanades, N. Bion, T. Le Mercier and V. Harlé, Energy Fuels, 2013, 27, 6068–6078 CrossRef CAS.
- A. C. Gladen and J. H. Davidson, Sol. Energy, 2016, 139, 524–532 CrossRef CAS.
- C. Muhich, M. Hoes and A. Steinfeld, Acta Mater., 2018, 144, 728–737 CrossRef CAS.
- J. R. Scheffe, D. Weibel and A. Steinfeld, Energy Fuels, 2013, 27, 4250–4257 CrossRef CAS.
- A. H. McDaniel, E. C. Miller, D. Arifin, A. Ambrosini, E. N. Coker, R. O'Hayre, W. C. Chueh and J. Tong, Energy Environ. Sci., 2013, 6, 2424–2428 CAS.
- A. Demont, S. Abanades and E. Beche, J. Phys. Chem. C, 2014, 118, 12682–12692 CAS.
- C.-K. Yang, Y. Yamazaki, A. Aydin and S. M. Haile, J. Mater. Chem. A, 2014, 2, 13612–13623 CAS.
- A. Demont and S. Abanades, RSC Adv., 2014, 4, 54885–54891 RSC.
- A. Demont and S. Abanades, J. Mater. Chem. A, 2015, 3, 3536–3546 CAS.
- A. H. Bork, M. Kubicek, M. Struzik and J. L. M. Rupp, J. Mater. Chem. A, 2015, 3, 15546–15557 CAS.
- M. E. Galvez, R. Jacot, J. Scheffe, T. Cooper, G. Patzke and A. Steinfeld, Phys. Chem. Chem. Phys., 2015, 17, 6629–6634 RSC.
- F. He and F. Li, Energy Environ. Sci., 2015, 8, 535–539 CAS.
- L. Wang, M. Al-Mamun, Y. L. Zhong, L. Jiang, P. Liu, Y. Wang, H. G. Yang and H. Zhao, Sustainable Energy Fuels, 2017, 1, 1013–1017 CAS.
- M. Orfila, M. Linares, R. Molina, J.-A. Botas, R. Sanz and J. Marugan, Int. J. Hydrogen Energy, 2016, 41, 19329–19338 CrossRef CAS.
- M. Ezbiri, M. Takacs, B. Stolz, J. Lungthok, A. Steinfeld and R. Michalsky, J. Mater. Chem. A, 2017, 5, 15105–15115 CAS.
- R. J. Carrillo and J. R. Scheffe, Sol. Energy, 2017, 156, 3–20 CrossRef CAS.
- M. M. Nair and S. Abanades, ChemistrySelect, 2016, 1, 4449–4457 CrossRef CAS.
- E. Konysheva and J. T. S. Irvine, Chem. Mater., 2009, 21, 1514–1523 CrossRef CAS.
- S. M. Haile, G. Staneff and K. H. Ryu, J. Mater. Sci., 2001, 36, 1149–1160 CrossRef CAS.
- N. Zakowsky, S. Williamson and J. T. S. Irvine, Solid State Ionics, 2005, 176, 3019–3026 CrossRef CAS.
- Y. Hou, M. Ding, S. Liu, S. Wu and Y. Lin, RSC Adv., 2014, 4, 5329–5338 RSC.
- C. Zhang, C. Wang, W. Zhan, Y. Guo, Y. Guo, G. Lu, A. Baylet and A. Giroir-Fendler, Appl. Catal., B, 2013, 129, 509–516 CrossRef CAS.
- R. P. Vasquez, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 14938–14941 CrossRef CAS.
- A. Nenning, A. K. Opitz, C. Rameshan, R. Rameshan, R. Blume, M. Havecker, A. Knop-Gericke, G. Rupprechter, B. Klotzer and J. Fleig, J. Phys. Chem. C, 2016, 120, 1461–1471 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00516d |
| This journal is © The Royal Society of Chemistry 2018 |