
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn N-heterocyclic carbene ligand promotes highly selective alkyne semihydrogenation with copper nanoparticles supported on passivated silica†
Nicolas
Kaeffer
 ,
Hsueh-Ju
Liu‡
,
Hung-Kun
Lo
,
Alexey
Fedorov
* and
Christophe
Copéret
*
,
Hsueh-Ju
Liu‡
,
Hung-Kun
Lo
,
Alexey
Fedorov
* and
Christophe
Copéret
*
ETH Zürich, Department of Chemistry and Applied Biosciences, Vladimir-Prelog-Weg 1-5, CH-8093, Zürich, Switzerland. E-mail: fedoroal@ethz.ch; ccoperet@ethz.ch
First published on 23rd May 2018
Abstract
We report a surface organometallic route that generates copper nanoparticles (NPs) on a silica support while simultaneously passivating the silica surface with trimethylsiloxy groups. The material is active for the catalytic semihydrogenation of phenylalkyl-, dialkyl- and diaryl-alkynes and displays high chemo- and stereoselectivity at full alkyne conversion to corresponding (Z)-olefins in the presence of an N-heterocyclic carbene (NHC) ligand. Solid-state NMR spectroscopy using the NHC ligand 13C-labeled at the carbenic carbon reveals a genuine coordination of the carbene to Cu NPs. The presence of distinct Cu surface environments and the coordination of the NHC to specific Cu sites likely account for the increased selectivity.
Introduction
N-Heterocyclic carbenes (NHCs) belong to a class of prominent ligands, especially in homogeneous catalysis,1–3 primarily due to their strong σ-donor properties4 that yield organometallic complexes with very stable metal–carbon bonds.2,5 NHC ligands have been utilized to improve the performance of numerous molecular catalysts in a broad range of applications such as olefin metathesis,6 cross-coupling7 and hydrogenation reactions,8–10 to mention just a few. NHCs also bind to the surfaces of metallic nanoparticles (NPs) and wafers,11–14 which was initially demonstrated for Au NPs15,16 and recently exploited in catalytic applications,17–21 such as electrocatalytic reduction of CO2.22,23 Another example is the use of Pd NPs decorated with NHC ligands for the Buchwald–Hartwig amination of aryl chlorides.24Selective semihydrogenation of alkynes to alkenes is an important process used in both industry and academia.25 The corresponding catalysts are typically based on Pd, among which the Lindlar catalyst26 is most frequently used, even though it relies on scarce and expensive Pd metal and requires toxic Pb additive to achieve high selectivity. Copper-based materials are known to catalyze semihydrogenation of alkynes under flow conditions.27,28 In this context, our group has recently reported that copper nanoparticles supported on a partially dehydroxylated silica (Cu/SiO2-700) efficiently catalyze batch semihydrogenation of alkynes in the presence of ligands (e.g. PCy3)29,30 with performances comparable to those of the Lindlar catalyst.31 However, with certain substrates these earth-abundant Cu catalysts still display limited selectivity at full alkyne conversion due to the secondary hydrogenation of formed alkenes.
We reasoned that ligand functionalization of NPs20,22–24,32–36 would improve chemoselectivity towards alkenes through competitive adsorption or poisoning of unselective sites by strong σ-donor ligands such as NHCs.29 However, free NHCs are strong bases (imidazolium/-ylidene couples typically have pKa values in the 20–29 range)37,38 and as such deprotonate the surface silanols of silica. In order to favor the selective coordination of an NHC ligand on the Cu sites, we use Surface Organometallic Chemistry (SOMC)39 with a tailored molecular precursor, [Cu4(HMDS)4] (HMDS = N(SiMe3)2) to synthesize Cu nanoparticles on passivated silica (that is silica where surface silanols are replaced by trimethylsilyloxy groups, SiO2-TMS).40,41 The resulting material (Cu/SiO2-TMS) is a highly active alkyne hydrogenation catalyst that becomes selective towards semihydrogenation by addition of a prototypical NHC ligand, IMes (IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene),42 through the binding of IMes to mostly Cu surface sites as revealed by solid-state NMR spectroscopy.
Results and discussion
Preparation of Cu/SiO2-TMS
The SOMC approach exploits the reactivity of [Cu4(HMDS)4] that was prepared on the gram scale from [Cu2(COD)2Cl2]43 (COD = 1,5-cyclooctadiene) and NaHMDS (39% yield, see ESI† for details).44 This Cu precursor reacts with SiO2-700 (2.1 equiv. Cu per surface![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–OH site) in THF at 60 °C (Scheme 1) to yield a material containing 2.12, 1.81, 0.29 and 0.26 wt% loadings of Cu, C, H and N, respectively, by elemental analysis.
Si–OH site) in THF at 60 °C (Scheme 1) to yield a material containing 2.12, 1.81, 0.29 and 0.26 wt% loadings of Cu, C, H and N, respectively, by elemental analysis.
 | ||
| Scheme 1 Synthetic route to 1 and 2. | ||
IR spectroscopy (Fig. 1) reveals that isolated silanols are fully consumed when [Cu4(HMDS)4] is contacted with SiO2-700 (disappearance of the sharp band at 3747 cm−1 with concomitant appearance of C–H stretching bands at 2902 and 2954 cm−1 associated with SiMe3 groups). The 1H, 13C and 29Si solid-state NMR spectra of the grafted material indicate the presence of SiMe3 groups in two different chemical environments (1H: 0.8 and 0.5 ppm, 13C: 7 and 0 ppm and 29Si: 2 and 15 ppm, Fig. S1†) corresponding to N(SiMe3)2 and OSiMe3, respectively.40
 | ||
| Fig. 1 FT-IR spectra of (a) [Cu4(HMDS)4], (b) SiO2-700, (c) [CuHMDS]/SiO2-TMS, and (d) Cu/SiO2-TMS (1) and (e) HR-TEM image and (f) particle size distribution of 1. | ||
These observations are consistent with the formation of isolated multinuclear Cu along with Si–OSiMe3 surface sites by grafting of [Cu4(HMDS)4] on SiO2-700via protonolysis, releasing HN(TMS)2 that further passivates remaining free silanols40,41 ([CuHMDS]/SiO2-TMS, Scheme 1). Treatment of [CuHMDS]/SiO2-TMS under a flow of H2 at 300 °C results in a color change from white to dark red and affords Cu/SiO2-TMS (1). The HR-TEM images of 1 show the presence of small, well dispersed Cu NPs with a narrow size distribution of 2.0 ± 0.6 nm (Fig. 1e and f). Elemental analysis of 1 indicates a Cu loading of 2.31 wt%, and H2 chemisorption gives ca. 0.07 mmol Cusurface g−1 in Cu surface sites (Fig. S2†). This loading is lower compared to that obtained using the [Cu5(Mes)5] precursor (5.5 wt%, 0.31 mmol Cusurface g−1),29 in agreement with the competing passivation of silanol sites by Me3Si groups that consequently limits the density of Cu sites upon grafting.
The IR spectrum of 1 displays a strong C–H stretching band at 2967 cm−1, along with a weak band at 3747 cm−1 that is associated with the presence of a small amount of residual Si–OH sites (ca. 10% with respect to initial silanols, Fig. 1d). NMR analysis (Fig. S1†) shows that 1 features a single set of peaks at 0.5 ppm (1H), 0 ppm (13C) and 15 ppm (29Si), pointing to a single chemical environment for SiMe3 groups. These observations are consistent with the formation of Cu NPs from Si–O–[Cun(HMDS)m] sites upon H2 treatment, regenerating surface silanols that are mostly directly passivated into Si–O–SiMe3 sites by released HN(SiMe3)2.
Activity in alkyne hydrogenation
Next, we recorded H2 consumption profiles during hydrogenation of a prototypical substrate 1-phenyl-1-propyne (S1) catalyzed by 1 (20 bar H2, 60 °C, 1.8 mol% Cutotal, 0.4 mol% Cusurface), both with or without additional ligands in the reaction media (Fig. 2). Two subsequent linear trends are observed in the H2 consumption profile for the unmodified catalyst 1 (Fig. 2c, black trace). The first linear slope of ca. 4.3 × 10−2 mol H2 L−1 h−1 (1.2 × 10−5 mol H2 L−1 s−1) at early stages is associated with hydrogenation of S1 to the (Z)-S12H. Following the full hydrogenation to S12H, the overhydrogenation of S12H to S14H occurs at a faster rate (second, steeper linear slope) of ca. 1.8 × 10−1 mol H2 L−1 h−1 (5.0 × 10−5 mol H2 L−1 s−1). GC analysis of the reaction mixture after 16 h confirms quantitative conversion of S1 to S14H (Fig. 2c, black, and Table 1, entry 1). The two-step behavior of the process likely originates from inhibition of the overhydrogenation by the starting alkyne until the latter gets depleted, and only then formation of S14H proceeds. The fast rate of overhydrogenation at high conversion is thus responsible for the quick deterioration of chemoselectivity in the desired cis-olefinic product. | ||
Fig. 2 Hydrogenation of alkynes with catalyst 1: (a) General reaction scheme, (b) substrate scope, (c) H2 consumption profiles for hydrogenation of 1-phenyl-1-propyne (S1, 400 μmol) at 20 bar, 60 °C in toluene over unmodified 1 (black, 1.8 mol% Cutotal) or in the presence of IMes (red) or PCy3 (blue) using a ligand/S1 molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, and (d) selectivity to (Z)-S12H among hydrogenation products S12H and S14H (selectivity[(Z)-S12H/S12H+4H]) at the final time (16 h). :50, and (d) selectivity to (Z)-S12H among hydrogenation products S12H and S14H (selectivity[(Z)-S12H/S12H+4H]) at the final time (16 h). | ||
| Entry | Ligand | L/S1 ratio | Sel. S12H (Z/E) (%) | Sel. S14H (%) |
|---|---|---|---|---|
| 1 | — | — | <1 | >99 |
| 2 | IMes | 1:50 |
97 (99:1) |
3 |
| 3 | IMes | 1:100 |
95 (98:2) |
5 |
| 4 | IMes | 1:500 |
12 (58:42) |
88 |
| 5 | PCy3 | 1:50 |
66 (89:11) |
34 |
Introducing 2 mol% of IMes (ca. 1:6 Cusurface/IMes ratio) to the catalytic mixture does not significantly change the initial rate of hydrogenation of 1-phenyl-1-propyne, as follows from a very similar slope in the H2 uptake profile (5.0 × 10−2 mol H2 L−1 h−1 and 1.4 × 10−5 mol H2 L−1 s−1; Fig. 2c, red trace). These unaltered rates suggest no poisoning of catalytic sites by IMes binding. However, the overhydrogenation to S14H is fully inhibited in the presence of IMes and the chemoselectivity to (Z)-S12H remains at 97% even after 16 h of reaction (Fig. 2c, red, and Table 1, entry 2). The high (Z)-S12H chemoselectivity is also retained at a lower IMes/S1 ratio of 1:100 (Table 1, entry 3 and Fig. S3†). Furthermore, GC analysis using an internal standard reveals only minor amounts of unidentified side products (1%, Table S1,† entry 2), possibly oligomers, confirming the very high overall selectivity to (Z)-S12H with added IMes.
Hot filtration experiments showed no further conversion of S1 after removal of catalysts from the reaction mixture with or without added IMes, thereby demonstrating that no leaching of copper occurs, consistent with our previous observations.29
Control experiments reveal that while (Z)-S12H is readily hydrogenated by 1, introducing IMes (2 mol% with respect to (Z)-S12H) completely shuts down the conversion of the alkene (Table S2†). This result clearly demonstrates that the NHC ligand inhibits hydrogenation of (Z)-S12H to S14H, thereby avoiding overhydrogenation. For comparison, when IMes is replaced by tricyclohexylphosphine (2 mol% PCy3), overhydrogenation is not completely hindered, thus compromising the selectivity, in particular at high conversion (Fig. 2c and d, blue traces, and Tables 1, entry 5, and S2†).
To illustrate that IMes generally improves selectivity for semihydrogenation, we submitted several additional phenylalkyl-, dialkyl- and diarylalkynes45 (S2–S7, Fig. 2b) to hydrogenation over 1 with or without added IMes. We optimized conditions (1:50 IMes/substrate ratio, 20 bar, 60 °C, 24 h, 9 mol% Cutotal, 2 mol% Cusurface) to ensure full conversion of the starting alkyne, which was successfully achieved for all substrates except S7 (vide infra, Table 2). At this extended reaction time and significantly higher catalyst loading, S1 retains good selectivity to S12H (88%) in the presence of IMes that is however completely lost due to S14H (>99%) when IMes is not added (Table 2, entries 1–2). Being structurally related to S1, 1-phenyl-1-hexyne (S2) likewise displays high selectivity for semihydrogenation to S22H (91%) only in the presence of IMes, whereas complete overhydrogenation to the corresponding alkane is observed using pristine 1; both reactions proceed without detected side-products (Table 2, entries 3–4). The selectivity to the (Z)-olefin is very high for the aliphatic alkyne S3 (99% to S32H, >99:1 Z/E ratio) when IMes is added to the reaction mixture, in contrast to what is observed in the absence of the ligand, where lower Z selectivity (Z/E = 91:9) and substantial by-product formation occur (17%), presumably due to oligomerization (Table 2, entries 5–6).
:50 (see ESI for details)
| Entry | Substrate | Ligand | Conv. (%) | Sel. S2H (%) | S 2H Z/E ratio | Sel. others (S4H) (%) |
|---|---|---|---|---|---|---|
| a Mass balance indicates the formation of undetected side products, likely oligomers. | ||||||
| 1 | S1 | — | >99 | <1 | — | >99 (>99) |
| 2 | S1 | IMes | >99 | 88 | 90:10 |
<1 (—) |
| 3 | S2 | — | >99 | <1 | — | >99 (>99) |
| 4 | S2 | IMes | >99 | 91 | 91:9 |
9 (9) |
| 5 | S3 | — | >99 | 83 | 91:9 |
17 (<1)a |
| 6 | S3 | IMes | >99 | 99 | >99:1 |
1 (<1)a |
| 7 | S4 | — | >99 | <1 | — | >99 (>99) |
| 8 | S4 | IMes | >99 | >99 | 97:3 |
<1 (—) |
| 9 | S5 | — | >99 | <1 | — | >99 (>99) |
| 10 | S5 | IMes | >99 | >99 | 97:3 |
<1 (—) |
| 11 | S6 | — | >99 | 28 | 48:52 |
72 (72) |
| 12 | S6 | IMes | >99 | 96 | 98:2 |
4 (4) |
| 13 | S7 | — | 79 | >99 | 99:1 |
<1 |
| 14 | S7 | IMes | 66 | >99 | 99:1 |
<1 |
The semihydrogenation of diphenylacetylenes S4 and S5 in the presence of IMes provides respective (Z)-olefins in very high yields (>96%); in contrast, complete overhydrogenation to the corresponding diphenylethanes occurs when IMes is not added (>99% selectivity, Table 2, entries 7–10). The chloro-derivative S6 yields mostly S64H (72% selectivity) over the unmodified catalyst 1 while S62H is obtained with 28% selectivity. Interestingly, S62H formed with pristine 1 is roughly an equal mixture of Z- and E-isomers (Table 2, entry 11). Addition of the IMes ligand alleviates low chemo- and stereoselectivities, yielding Z-stilbene S62H in 93% yield (Table 2, entry 12). The bromo-derivative S7 does not reach full conversion both with or without added IMes (66 and 79% conversion, respectively) and features high selectivity to (Z)-S72H in both cases (>99%, entries 13–14). This high chemoselectivity to (Z)-S72H likely originates from the remaining alkyne S7, consistent with the inhibition of the overhydrogenation by the starting alkyne, as was also reported previously.29 We note that both halogen-containing substrates S6 and S7 react slower over catalyst 1, possibly due to the interaction of the Cu surface with the halogen substituent. The latter effect would also be consistent with the high fraction of a chloro-stilbene (E)-S62H formed with pristine catalyst 1, probably through a secondary isomerization of (Z)-S62H on a Cu surface that had possibly been modified by an interaction with the Cl-substituent (Table 2, entry 11). Overall, these results demonstrate that the enhancement in selectivity provided by IMes can be extended to phenylalkyl, dialkyl and diaryl internal alkynes. These findings further underline the impressive effect of IMes on inhibiting the undesired overhydrogenation of internal olefins and also hindering side reactions such as oligomerization and olefin isomerization, especially at elevated temperature (60 °C).29
Characterizing the ligand–particle interaction
To understand the origin of the improved chemoselectivity, we studied the nature of the IMes–Cu NP interaction at the molecular level using solid-state NMR. Contacting 1 with a toluene solution of IMes*, 13C-labeled at the carbenic carbon, followed by washing of the resulting material with toluene and pentane and drying under vacuum yields IMes*–Cu/SiO2-TMS (2*, the labeled analogue of 2, Scheme 1). Elemental analysis of 2* gives 1.82, 3.37, 0.48 and 0.44 wt% Cu, C, H and N, respectively, consistent with the immobilization of IMes* on Cu/SiO2-TMS (Table S3†). Elemental analysis data allow estimating the Cusurface/IMes ratio to be ca. 1:1.4, suggesting concomitant binding of IMes* on both Cu NPs and the support. The IR spectrum of 2 contains bands reminiscent of the NHC ligand, confirming adsorption of IMes onto 1 (Fig. S4†). Exposure of 1 to 33 mbar of CO (Fig. S5†) gives a broad IR band between 2000–2200 cm−1 that can be decomposed into 3 main components centered at 2119, 2095 and 2055 cm−1. Repeating the experiment with 2 (Fig. S5†) produces CO bands that are less intense, possibly because its adsorption is hindered by the bulky NHC ligand, as already observed with PCy3-functionalized non-passivated Cu/SiO2-700.29 Moreover, the feature at 2119 cm−1 disappears whereas a new band appears at 2004 cm−1, resulting in an overall red shift of CO adsorption modes. This red shift points to a stronger back-donation to CO, consistent with more electron-rich Cu NPs as expected from binding of an IMes ligand to the Cu surface. Furthermore, X-ray absorption near edge structure (XANES) spectroscopy of 1 and 2 demonstrates a slight change in the Cu-K white line from 8996.5 to 8996.7 eV that also supports binding of IMes on Cu NPs (Fig. S6†).
NMR spectroscopy is particularly instrumental in elucidating the coordination of ligands, including NHCs, on transition-metal NPs.18,19,22,24,46–48 The 13C direct excitation spectrum of 2* (Fig. 3b) reveals a major, broad peak centered at 178 ppm and a shoulder at 189 ppm, which are also present in the 13C cross-polarization (CP) spectrum (Fig. 3c). In contrast, these peaks disappear in the 13C CP spectrum of 2 (Fig. 3e) and can thus be attributed to 13C-labeled carbons in 2*. In addition, the intensity of the peak at 178 ppm relative to that of TMS groups of the support (−2 ppm) increases in the direct excitation spectrum compared to the CP one (Fig. S7b and c†), pointing to carbon atoms not directly bonded to protons. With these observations, we assign the peak at 178 ppm to (13C-labeled) carbenic carbons coordinated to Cu NPs. This assignment is further supported by the upfield shift of this peak compared to the carbenic carbon in free IMes* (219 ppm, Fig. 3a)49 and by the similarity of the observed shifts to values reported for NHC bound to Cu sites in [Cu(IMes)(L)] molecular complexes.50 In the same ppm range, the shoulder at 189 ppm is also attributed to Cu-bound carbenic carbons, likely in a different environment. The relative intensity of this peak is similar in the direct excitation and CP spectra (Fig. S7b and c†), suggesting the presence of protons in the vicinity of the carbene ligand, possibly associated with Cu sites closer to the interface with the SiO2-TMS support. Alternative interpretations are that distinct resonances at 178 and 189 ppm arise from IMes bound on (111) and (200) facets, which are expected to be present on Cu nanoparticles,51 or from high (terraces) and low (corners or edges) coordination sites present on Cu NPs.
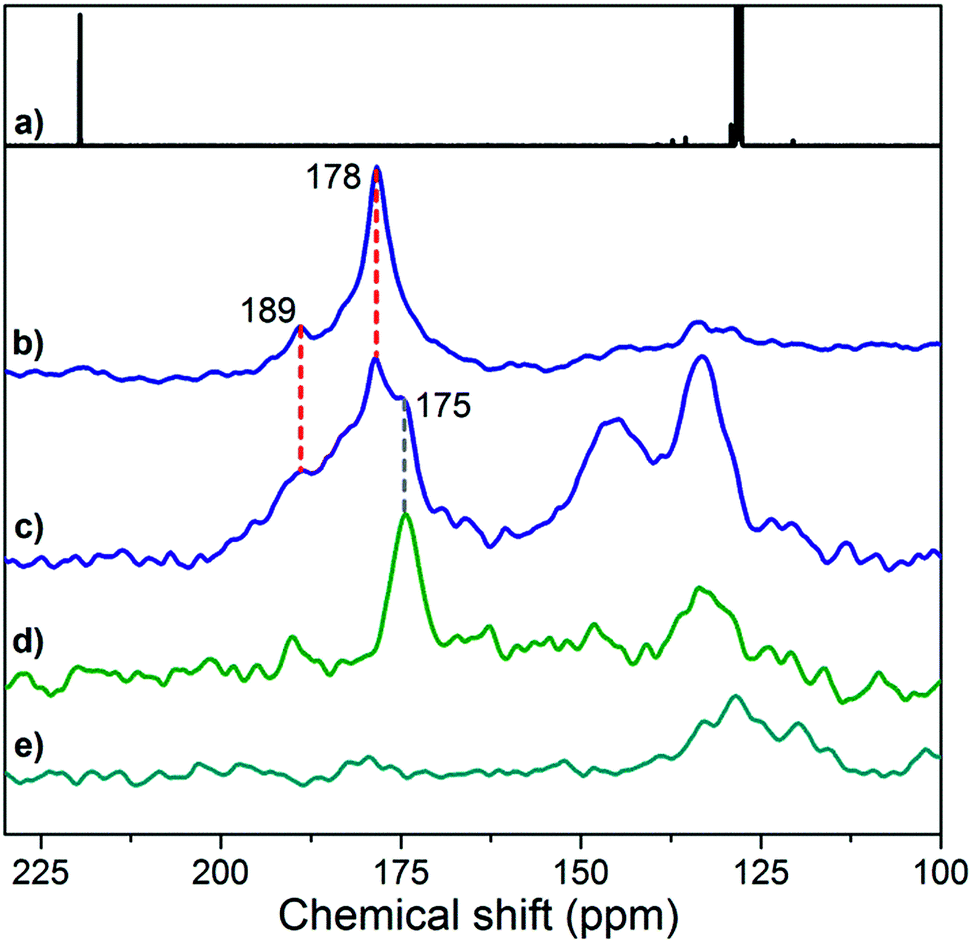 | ||
| Fig. 3 (a) 13C NMR solution-state spectrum of free IMes* (C6D6, 75 MHz); (b) 13C NMR solid-state HPDEC spectrum of 2* (30000 scans); 13C NMR solid-state CP-MAS spectra of (c) 2* (90000 scans), (d) IMes*/SiO2-TMS (27513 scans) and (e) 2 (90000 scans). Broader spectral windows are provided in Fig. S7 and S8† and 1H NMR spectra for IMes* and 2* are given in Fig. S9.† | ||
The peak at 175 ppm in the CP spectrum of 2* (Fig. 3c) is attributed to the 13C-labeled carbenic carbon of IMes* in interaction with the support, as this peak is also found for the fully passivated SiO2-TMS support contacted with IMes* (IMes*/SiO2-TMS, Fig. 3d and S7e†) and it may be due to the carbene coordinated to Si atoms52 in TMS groups or siloxane bridges. Peaks at 133 and 145 ppm in the CP spectrum of 2* (Fig. 3c) are also observed for non-passivated SiO2-700 treated with IMes* (Fig. S7f†); they are assigned to 13C-labeled imidazolium species formed on the support upon deprotonation of residual silanols by IMes*. The observation of peaks attributed to surface species on the support itself is also consistent with elemental analysis of 2*, which shows the presence of an excess of the ligand per available surface Cu atom.
Collectively, these results prove the immobilization of IMes in 2* with a partial coverage of the support and actual ligand–particle interaction through genuine coordination of the carbenic carbon to the copper particle.
Conclusion
In conclusion, we report the controlled synthesis of small Cu NPs supported on Me3Si-passivated SiO2-700 through SOMC. The supported Cu NPs are highly active for the hydrogenation of alkynes, and introduction of an NHC ligand greatly improves the selectivity of these particles for semihydrogenation of phenylalkyl, dialkyl and diaryl internal alkynes generating the corresponding cis-olefins with very high selectivities (>95%) at full conversion. This increased selectivity likely arises from the binding of the IMes ligand to Cu NPs, in a genuine coordination which has been confirmed by 13C NMR spectroscopy. This work shows that inexpensive and readily available supported Cu nanoparticles can be turned into highly selective catalysts by choosing appropriate coordinating ligands to modulate their activity and selectivity. Our group is currently exploring this research direction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Scientific Equipment Program of ETH Zürich and the SNSF (R'Equip grant 206021_150709/1) for financial support of the high throughput catalyst screening facility (HTE@ETH). N. K. acknowledges support from the ETHZ Postdoctoral Fellowship Program and from the Marie Curie Actions for People COFUND Program. H.-J. L. was partially funded by CCEM. A. F. thanks the Holcim Stiftung for a habilitation fellowship. The development of supported Cu catalysts was also partially funded by the SCCER Heat and Energy Storage. The authors thank W.-C. Liao and C. Gordon for their assistance with the NMR measurements, Dr K. Larmier and Dr T. Margossian for XAS measurements and Dr F. Krumeich (ScopeM) for TEM images.Notes and references
- J. C. Lin, R. T. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef PubMed
.
- C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef PubMed
- T. Droge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef PubMed
- W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162–2187 CrossRef
- G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef PubMed
- R. D. J. Froese, C. Lombardi, M. Pompeo, R. P. Rucker and M. G. Organ, Acc. Chem. Res., 2017, 50, 2244–2253 CrossRef PubMed
- E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239–2246 CrossRef
- D. Zhao, L. Candish, D. Paul and F. Glorius, ACS Catal., 2016, 6, 5978–5988 CrossRef
- J. W. Sprengers, J. Wassenaar, N. D. Clement, K. J. Cavell and C. J. Elsevier, Angew. Chem., Int. Ed., 2005, 44, 2026–2029 CrossRef PubMed
- A. V. Zhukhovitskiy, M. J. MacLeod and J. A. Johnson, Chem. Rev., 2015, 115, 11503–11532 CrossRef PubMed
- K. Salorinne, R. W. Y. Man, C. H. Li, M. Taki, M. Nambo and C. M. Crudden, Angew. Chem., Int. Ed., 2017, 56, 6198–6202 CrossRef PubMed
- C. M. Crudden, J. H. Horton, I. I. Ebralidze, O. V. Zenkina, A. B. McLean, B. Drevniok, Z. She, H. B. Kraatz, N. J. Mosey, T. Seki, E. C. Keske, J. D. Leake, A. Rousina-Webb and G. Wu, Nat. Chem., 2014, 6, 409–414 CrossRef PubMed
- G. Wang, A. Ruhling, S. Amirjalayer, M. Knor, J. B. Ernst, C. Richter, H. J. Gao, A. Timmer, H. Y. Gao, N. L. Doltsinis, F. Glorius and H. Fuchs, Nat. Chem., 2017, 9, 152–156 CrossRef PubMed
- J. Vignolle and T. D. Tilley, Chem. Commun., 2009, 7230–7232 RSC
- E. C. Hurst, K. Wilson, I. J. S. Fairlamb and V. Chechik, New J. Chem., 2009, 33, 1837 RSC
- C. Richter, K. Schaepe, F. Glorius and B. J. Ravoo, Chem. Commun., 2014, 50, 3204–3207 RSC
- L. M. Martinez-Prieto, A. Ferry, L. Rakers, C. Richter, P. Lecante, K. Philippot, B. Chaudret and F. Glorius, Chem. Commun., 2016, 52, 4768–4771 RSC
- J. B. Ernst, S. Muratsugu, F. Wang, M. Tada and F. Glorius, J. Am. Chem. Soc., 2016, 138, 10718–10721 CrossRef PubMed
- F. Novio, D. Monahan, Y. Coppel, G. Antorrena, P. Lecante, K. Philippot and B. Chaudret, Chem. –Eur. J., 2014, 20, 1287–1297 CrossRef PubMed
- K. V. Ranganath, J. Kloesges, A. H. Schafer and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 7786–7789 CrossRef PubMed
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong, J. A. Reimer, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef PubMed
- Z. Cao, J. S. Derrick, J. Xu, R. Gao, M. Gong, E. M. Nichols, P. T. Smith, X. Liu, X. Wen, C. Copéret and C. J. Chang, Angew. Chem., Int. Ed., 2018, 57, 4981–4985 CrossRef PubMed
- J. B. Ernst, C. Schwermann, G. I. Yokota, M. Tada, S. Muratsugu, N. L. Doltsinis and F. Glorius, J. Am. Chem. Soc., 2017, 139, 9144–9147 CrossRef PubMed
- G. Vilé, D. Albani, N. Almora-Barrios, N. López and J. Pérez-Ramírez, ChemCatChem, 2016, 8, 21–33 CrossRef
- H. Lindlar, Helv. Chim. Acta, 1952, 35, 446–450 CrossRef
- B. Bridier, M. A. G. Hevia, N. López and J. Pérez-Ramírez, J. Catal., 2011, 278, 167–172 CrossRef
- R. A. Koeppel, J. T. Wehrli, M. S. Wainwright, D. L. Trimma and N. W. Cant, Appl. Catal., A, 1994, 120, 163–177 CrossRef
- A. Fedorov, H. J. Liu, H. K. Lo and C. Copéret, J. Am. Chem. Soc., 2016, 138, 16502–16507 CrossRef PubMed
- O. G. Salnikov, H. J. Liu, A. Fedorov, D. B. Burueva, K. V. Kovtunov, C. Copéret and I. V. Koptyug, Chem. Sci., 2017, 8, 2426–2430 RSC
- C. Oger, L. Balas, T. Durand and J. M. Galano, Chem. Rev., 2013, 113, 1313–1350 CrossRef PubMed
- I. Schrader, S. Neumann, A. Šulce, F. Schmidt, V. Azov and S. Kunz, ACS Catal., 2017, 7, 3979–3987 CrossRef
- I. Schrader, S. Neumann, R. Himstedt, A. Zana, J. Warneke and S. Kunz, Chem. Commun., 2015, 51, 16221–16224 RSC
- F. Zaera, ACS Catal., 2017, 7, 4947–4967 CrossRef
- S. Kunz, Top. Catal., 2016, 59, 1671–1685 CrossRef
- J. L. Castelbou, A. Gual, E. Mercadé, C. Claver and C. Godard, Catal. Sci. Technol., 2013, 3, 2828 Search PubMed
- R. S. Massey, C. J. Collett, A. G. Lindsay, A. D. Smith and A. C. O'Donoghue, J. Am. Chem. Soc., 2012, 134, 20421–20432 CrossRef PubMed
- E. M. Higgins, J. A. Sherwood, A. G. Lindsay, J. Armstrong, R. S. Massey, R. W. Alder and A. C. O'Donoghue, Chem. Commun., 2011, 47, 1559–1561 RSC
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Nunez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed
- E. Oakton, G. Vile, D. S. Levine, E. Zocher, D. Baudouin, J. Perez-Ramirez and C. Copéret, Dalton Trans., 2014, 43, 15138–15142 RSC
- D. Gajan, K. Guillois, P. Delichere, J. M. Basset, J. P. Candy, V. Caps, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2009, 131, 14667–14669 CrossRef PubMed
- A. J. Arduengo, H. V. R. Dias, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1992, 114, 5530–5534 CrossRef
- K. M. Chi, H. K. Shin, M. J. Hampden-Smith, E. N. Duesler and T. T. Kodas, Polyhedron, 1991, 10, 2293–2299 CrossRef
- A. M. James, R. K. Laxman, F. R. Fronczek and A. W. Maverick, Inorg. Chem., 1998, 37, 3785–3791 CrossRef PubMed
- F. Chen, C. Kreyenschulte, J. Radnik, H. Lund, A.-E. Surkus, K. Junge and M. Beller, ACS Catal., 2017, 7, 1526–1532 CrossRef
- J. M. Asensio, S. Tricard, Y. Coppel, R. Andres, B. Chaudret and E. de Jesus, Angew. Chem., Int. Ed., 2017, 56, 865–869 CrossRef PubMed
- P. Lara, L. M. Martínez-Prieto, M. Roselló-Merino, C. Richter, F. Glorius, S. Conejero, K. Philippot and B. Chaudret, Nano-Struct. Nano-Objects, 2016, 6, 39–45 CrossRef
- P. Lara, O. Rivada-Wheelaghan, S. Conejero, R. Poteau, K. Philippot and B. Chaudret, Angew. Chem., Int. Ed., 2011, 50, 12080–12084 CrossRef PubMed
- D. Tapu, D. A. Dixon and C. Roe, Chem. Rev., 2009, 109, 3385–3407 CrossRef PubMed
- O. Santoro, A. Collado, A. M. Slawin, S. P. Nolan and C. S. Cazin, Chem. Commun., 2013, 49, 10483–10485 RSC
- K. Larmier, S. Tada, A. Comas-Vives and C. Copéret, J. Phys. Chem. Lett., 2016, 7, 3259–3263 CrossRef PubMed
- F. Bonnette, T. Kato, M. Destarac, G. Mignani, F. P. Cossio and A. Baceiredo, Angew. Chem., Int. Ed., 2007, 46, 8632–8635 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods, additional catalysis data and IR, NMR and XAS spectra. See DOI: 10.1039/c8sc01924j |
| ‡ Present address: Department of Applied Chemistry, National Chiao Tung University, 1001 University Road, Hsinchu, 30010, Taiwan. |
| This journal is © The Royal Society of Chemistry 2018 |