
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative β-C–H sulfonylation of cyclic amines†
R. J.
Griffiths
 ab,
W. C.
Kong
ac,
S. A.
Richards
a,
G. A.
Burley
b,
M. C.
Willis
*c and
E. P. A.
Talbot‡
*a
ab,
W. C.
Kong
ac,
S. A.
Richards
a,
G. A.
Burley
b,
M. C.
Willis
*c and
E. P. A.
Talbot‡
*a
aGlaxoSmithKline Medicines Research, Gunnels Wood Road, Stevenage, Hertfordshire SG1 2NY, UK
bDepartment of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK
cDepartment of Chemistry, University of Oxford, CRL Mansfield Road, Oxford, OX1 3TA, UK. E-mail: michael.willis@chem.ox.ac.uk
First published on 22nd January 2018
Abstract
A transition metal-free strategy for the dehydrogenative β-sulfonylation of tertiary cyclic amines is described. N-Iodosuccinimide facilitates regioselective oxidative sulfonylation at C–H bonds positioned β to the nitrogen atom of tertiary amines, installing enaminyl sulfone functionality in cyclic systems. Mild reaction conditions, broad functional group tolerance and a wide substrate scope are demonstrated. The nucleophilic character of the enaminyl sulfone is harnessed, demonstrating potential application for scaffold diversification.
Introduction
Aliphatic azacycles are essential motifs in drug discovery, with 59% of unique small-molecule drugs approved by the FDA containing at least one nitrogen heterocycle.1a Of these, the piperidine motif is the most prevalent nitrogen ring-system, highlighting the importance of this heterocycle in small-molecule drug discovery.1 Simple piperidines are readily available, hence methods for the straightforward late-stage diversification of this ring-system, ideally exploiting C–H functionalization, are valuable tools for medicinal chemistry (Scheme 1a).2 The majority of reported methods for the C–H functionalization of saturated nitrogen heterocycles have primarily focused on activation of the position α to the nitrogen atom, with methods based on direct lithiation, as well a catalytic C–H bond cleavage being described.3 Sanford has recently reported an elegant process for the palladium-catalyzed transannular C–H arylation of piperidines (γ-functionalization),4a and Bull has reported the palladium catalyzed C-3 arylation of proline derivatives (β-functionalization);4b it should be noted that both of these systems rely on a pre-installed directing group. Additional reports of functionalization remote to the nitrogen atom of cyclic amines are somewhat less well-precedented.4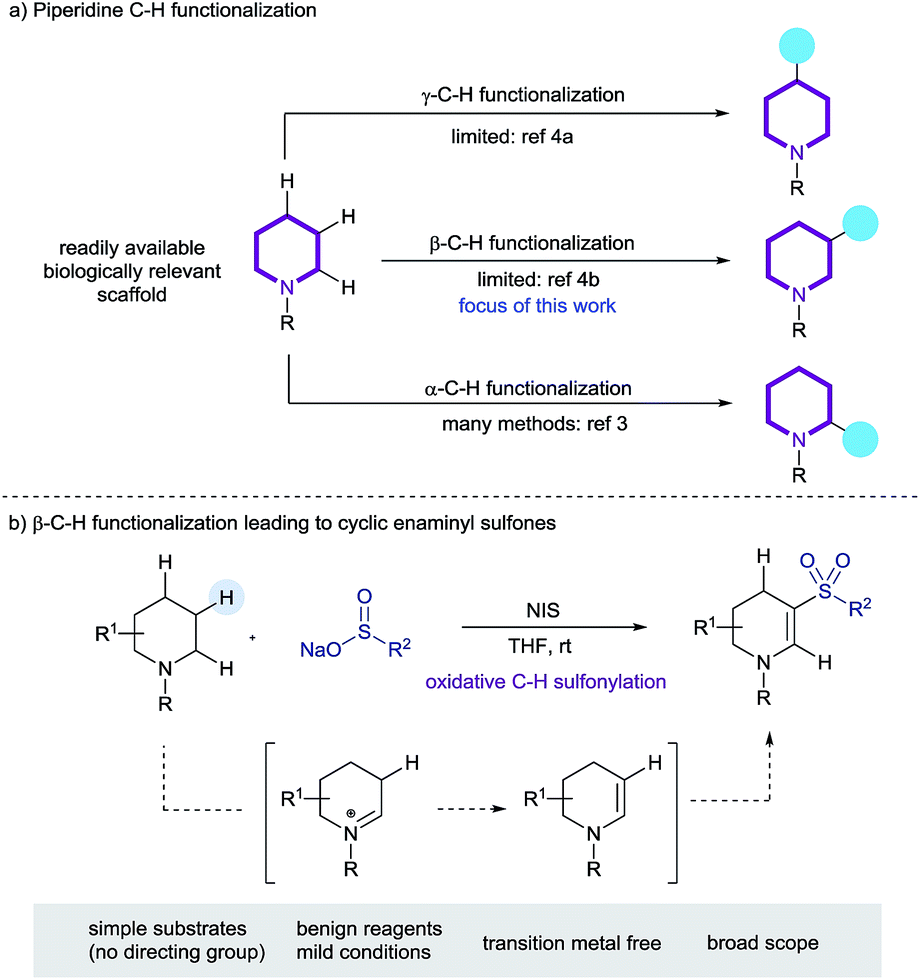 | ||
| Scheme 1 (a) The C–H functionalization of piperidines, and (b) this work: the preparation cyclic enaminyl sulfones. | ||
Sulfones are privileged functional groups in the pharmaceutical and agrochemical industries,5 and serve as versatile intermediates for organic synthesis.6 Recent methods for the preparation of sulfones have focused on the utilization of higher valent sulfur reagents in order to avoid oxidative transformations.7 Sulfinate salts have been employed for the direct formation of vinyl and aryl sulfones,8 and methods based on the trapping of sulfur dioxide have also been exploited.9 The varied methods available for sulfone preparation, together with their proven worth in medicinal chemistry, make them ideal functional groups to install using a C–H functionalization approach.
We recently described the iodine-mediated conversion of cyclic amines to lactams, in what corresponds to an α-C–H functionalization process.10 Iodine has also been used, along with an excess of peroxide co-oxidant, for the formation of enaminyl sulfones using simple acyclic amines and sulfinates salts as starting materials.11 A related, visible light mediated transformation employing air sensitive sulfonyl chlorides and a large excess of amine substrate (>4 eq.) has also been reported by Zheng and Zhang, but in both cases the product enaminyl sulfones were obtained in only poor yields and selectivities.12 Both of these reports focus mostly on acyclic amine substrates and propose that an intermediary enamine attacks an electrophilic sulfonyl species. Our previous work showed evidence of proceeding via an iminium/enamine pathway, thus we speculated that formation of cyclic enaminyl sulfones from cyclic amines and sulfinates should be feasible under oxidative iodine conditions, and that such a reaction would provide a valuable transformation for the β-C–H functionalization of piperidines (Scheme 1b). In this article we report the successful realization of this goal, and use the installed functionality as a unique nucleophile for wider functionalisation.
Results and discussion
Important objectives at the start of our study were to define reaction conditions that would deliver efficient and selective reactions with good functional group tolerance. In particular, we were keen to avoid the use of tert-butyl hydroperoxide as a reagent, and also targeted ambient temperature reactions. Capitalizing on our prior report of iodine-mediated α-C–H oxidation of amines, which proceeded at room temperature, we began our reaction optimization using iodine-based reagents. N-Benzyl piperidine 1a was used as a model substrate and was combined with sodium p-tolylsulfinate in our optimization studies. A robust screen of conditions was undertaken, surveying the parameters of solvent, concentration, temperature, type of halo-oxidant and stoichiometry of oxidant and sulfinate salt. The optimal conditions were identified, and THF proved to be the optimal solvent for this transformation, and N-iodosuccinimide (NIS) the optimal oxidant (Table 1).|
|
||||
|---|---|---|---|---|
| Entry | Equiv. p-TolSO2Na | Oxidant | Reaction conditions | % 2ab |
| a Reaction conditions: 1a (1.0 eq.), oxidant (4.0 eq.), solvent, 0.5 h, then p-TolSO2Na, solvent (0.063 M), rt, 2 h. b % conversion to 2a was measured by 1H NMR analysis of the crude material against 3,4,5-trichloropyridine as an internal standard. c “actv” refers to the pre-stirring of oxidant with 1a. d THF contained 250 ppm BHT radical inhibitor. e THF was inhibitor-free. f p-TolSO2Li was used as the sulfinate salt instead. p-Tol = para-tolyl. | ||||
| 1 | 3 | NIS | RT, 0.5 h actvc, 3 h, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 THFd:water, 5 eq. NaHCO3 :1 THFd:water, 5 eq. NaHCO3 |
43 |
| 2 | 3 | NIS | RT, 0.5 h actv, 3 h, 2:1 DMSO:water, 5 eq. NaHCO3 |
38 |
| 3 | 3 | NIS | RT, 0.5 h actv, 2 h, DCM | 27 |
| 4 | 3 | NIS | RT, 0.5 h actv, 2 h, DMSO | 60 |
| 5 | 3 | I2 | RT, 0.5 h actv, 2 h, DMSO | Trace |
| 6 | 3 | ICl | RT, 0.5 h actv, 2 h, DMSO | — |
| 7 | 3 | NIS | RT, 0.5 h actv, 2 h, DMSO, N2, dark | 81 |
| 8 | 3 | NIS | RT, 0.5 h actv, 2 h, THFe, N2, dark | 95 |
| 9 | 3 | NIS | RT, 0.5 h actv, 2 h, 2-MeTHF, N2, dark | 65 |
| 10 | 1.5 | NIS | RT, 0.5 h actv, 2 h, THF , N 2 , dark | 90 |
| 11 | 1.5f | NIS | RT, 0.5 h actv, 2 h, THFe, N2, dark | 71 |
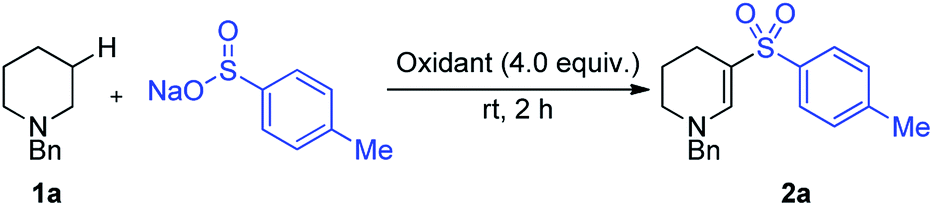
In contrast to our previous work on the iodine-mediated formation of lactams, water and base were not found to be necessary for the formation of 2a, with anhydrous DMSO providing the best conversion to 2a (entry 4). Molecular iodine and iodine monochloride (entries 5 and 6) were significantly inferior oxidants than NIS. Shielding the reaction from light and oxygen was found to be crucial to achieving more reproducible results, and led to improved conversion to 2a (entry 7). Finally, using THF as the solvent enabled a further increase in the formation of 2a (entry 8), and also provided conditions that were amenable to reducing the amount of the more expensive sulfinate salt to only 1.5 equivalents (entry 10), with the reaction proceeding to 90% conversion. A slight decrease in conversion to 2a (entry 11, 71%) was observed when using the lithium sulfinate salt, suggesting an importance the less tightly bound anion in the sodium sulfinate increases reactivity. However, the lithium sulfinate still proceeded well, which provides wider applicability for sulfinates synthesized via a lithiation protocol. Full details of the reaction optimization can be found in the ESI (Table S1†).
With the optimized conditions in hand, the scope of the reaction with respect to variation of sodium sulfinate salt was explored (Table 2). Electron-rich (2b), electron-poor (2c) and halide substituted (2d–f) aryl sulfinates provided access to enaminyl sulfones in good to excellent conversions and yields (68–98%). This provides an opportunity for subsequent diversification using the pre-installed halide functional group for the development of molecular libraries of biologically relevant molecules. Meta- and ortho-substitution on the aryl ring was also tolerated well (2g and h, 91% and 73%, respectively), demonstrating good tolerance to steric crowding. Larger-scale reactions, performed on 5.42 mmol and 3.75 mmol of amine, delivered products 2a (75%) and 2g (70%), respectively, demonstrating the preparative utility of the method.

Heterocyclic sulfinates (2i–l) also performed well, although a lower conversion was observed for the 2-pyridyl sulfinate. Non-aryl sulfinates failed to provide the targeted enaminyl sulfones, with the only exceptions being the use of sodium cyclopropylsufinate and sodium styrylsulfinate, which provided enaminyl sulfones 2m and 2n in 68% and 47% yield, respectively.
The scope of amine component was evaluated next (Table 3). Variation of the electronics of the N-benzyl substituent was tolerated (4a, b), with no benzylic functionalization observed in either reaction. Sulfone 4c, featuring an arylmethylsulfide substituent, was obtained in 87% conversion, highlighting the tolerance of the reaction to oxidation-sensitive functional groups. Increasing the steric crowding around the nitrogen center was not detrimental to the reaction, and product 4d was isolated in 94% yield.
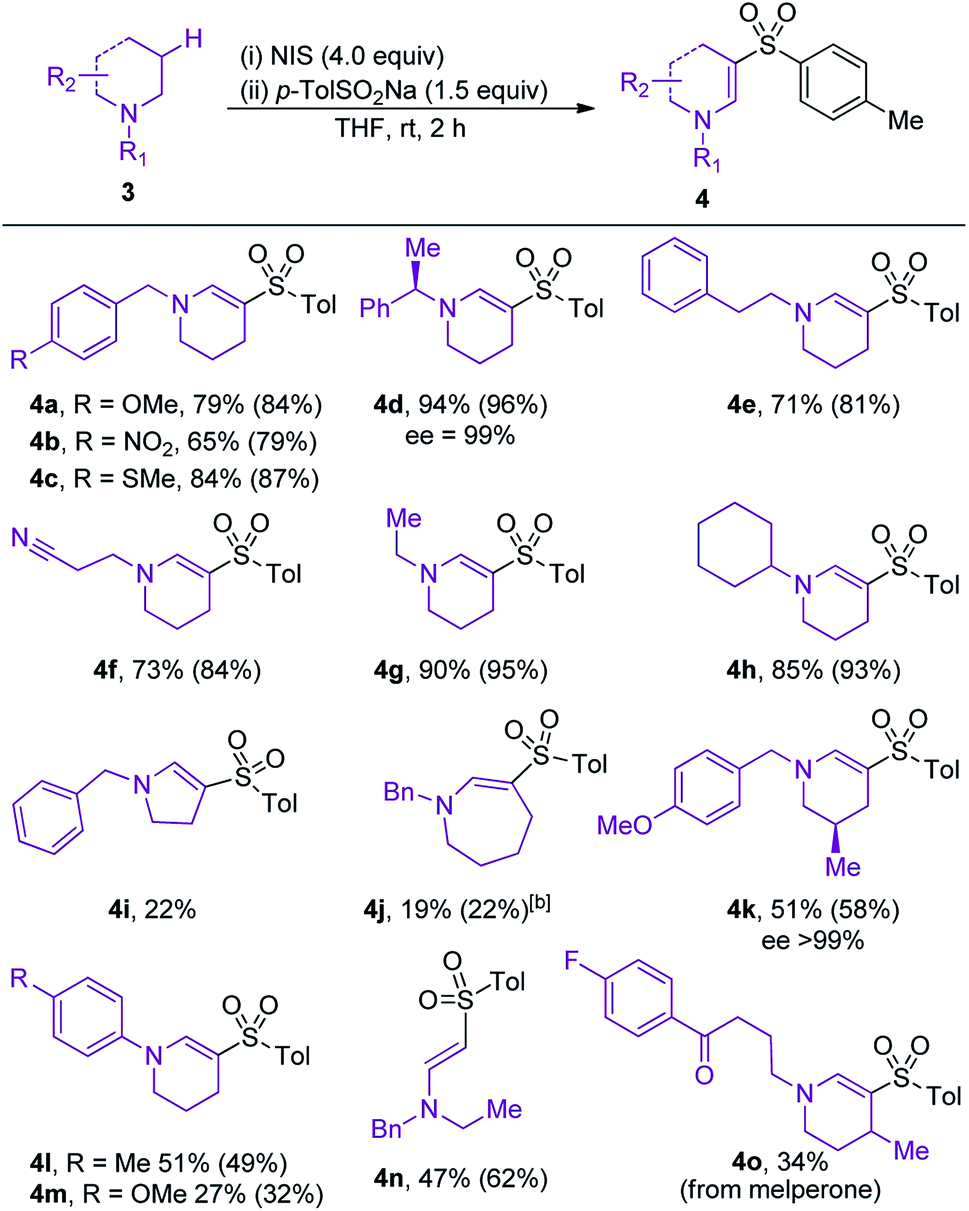
Sulfones 4e and 4f were obtained with high selectivities and yields, confirming the preference for endocyclic over exocyclic oxidation, and tolerance of a nitrile functional group. The reaction was also tolerant and selective for different N-alkyl substituents on the amine, with ethyl and cyclohexyl examples performing well (4g and h). Disappointingly, variation of the ring size of the cyclic amine (4i and j) was not tolerated well, with the five- and seven-membered amines providing only moderate yields of the desired enaminyl sulfones. The presence of a methyl-substituent on the framework of the piperidine ring steered sulfonylation to the less-hindered position, and provided sulfone 4k with high regioselectivity. N-Aryl amines performed moderately well, affording sulfones 4l and 4m in 49% and 32% conversion, respectively. The formation of sulfone 4n established that oxidative sulfonylation can be achieved for non-cyclic amines, and contrasts with our earlier iodine-mediated oxidative formation of amides, which was restricted to cyclic systems. The final example in Table 3 highlights the utility of the developed reaction for the late-stage C–H functionalization of medicinally relevant compounds; enaminyl sulfone 4o is derived from oxidative sulfonylation of melperone, a marketed atypical antipsychotic. The formation of 4o demonstrates how the developed reaction could be applied for the diversification of compound collections used in drug discovery.
As a preliminary investigation into the mechanism of the developed reaction we performed several control reactions (Table 4). The inclusion of BHT as an additive did not lead to appreciable inhibition of the reaction (entry 2). Catechol, however, resulted in a substantial drop in conversion, while sulfone 2a was not observed when TEMPO was added to the reaction (entries 3 and 4, respectively). Lack of inhibition by BHT despite inhibition occurring with the radical scavenger TEMPO has some precedent in related iodine/sulfinate reaction systems,8e thus a radical pathway could not be completely excluded at this stage.
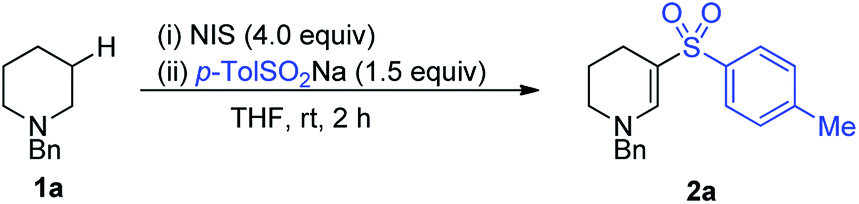
However, a radical clock experiment with 3p provided a mixture of 4pα and 4pβ, with no opening of the cyclopropyl ring observed (Scheme 2).
 | ||
| Scheme 2 Radical-clock experiment with 3p, indicating that the reaction does not proceed via a radical-based mechanism. Isolated yields shown; values in parentheses show conversion to product as measured by 1H NMR analysis of crude product mixture using an internal standard. Tol = para-tolyl. | ||
This result suggests that in fact the oxidative C–H sulfonylation reaction does not proceed via a radical-mediated pathway. The light and air-sensitivity of the reaction is indicative of formation of a sulfonyl iodide in situ, which are known to be unstable in the presence of light and oxygen.13 This accounts for the poorer results observed for aliphatic and hetereoaromatic sulfinate salts, as hetereoaromatic and alkyl sulfonyl halides have been known to be unstable.14 The inhibition of the reaction by known radical inhibitors is therefore proposed to arise as a result of reaction these additives accelerating the decomposition of the sulfonyl iodide. At this stage, the reaction is proposed to follow oxidation to an enamine, followed by nucleophilic attack of the sulfonyl iodide, though further studies are required.
Despite the combination of synthetically useful functional groups present in enaminyl sulfones, their use in synthesis has not been well explored.15 One reason for this is presumably the lack of convenient methods for their preparation. Accordingly, we set out to explore the utility of the cyclic enaminyl sulfone products obtained in this study as templates for further diversification (Scheme 3). Selective reduction of the enamine could be achieved under acidic silane conditions,16 affording saturated system 5 in an excellent 91% yield. Alternatively, hydrogenation over Pd/C using Zn/HCl in COware delivered sulfone 6 in comparable yield. Use of a flow hydrogenator enabled straightforward small-scale hydrogenation at high pressure; the use of 25 bar pressure enabled global hydrogenation to prepare piperidine 7 in which the N-benzyl group has been cleaved in 76% yield.
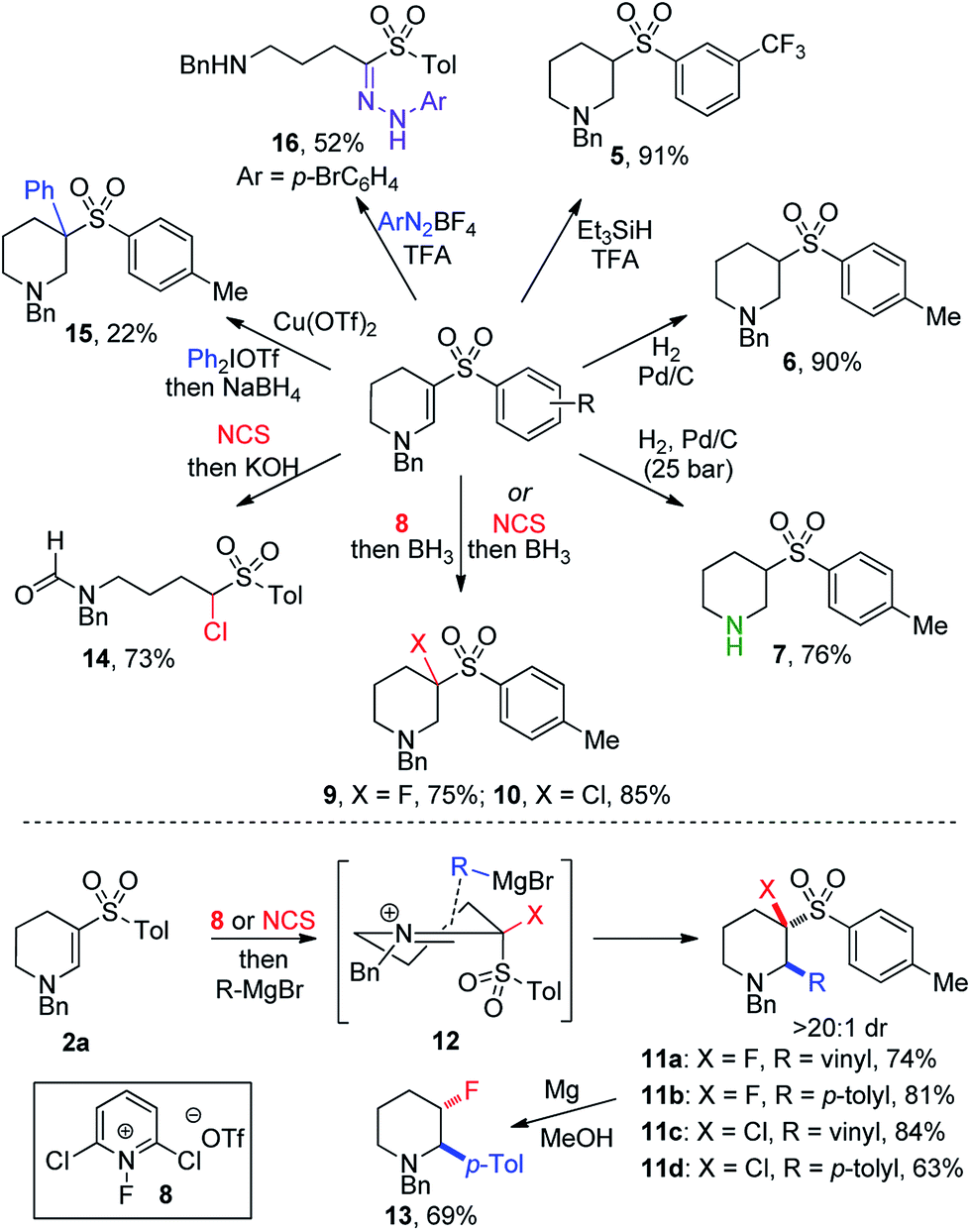 | ||
| Scheme 3 Cyclic enaminyl sulfones as templates for synthesis. Tol = para-tolyl. | ||
Incorporation of fluorine and chlorine atoms onto heterocyclic scaffolds is important for both modulating physiochemical properties,17 and for the introduction of synthetic handles for subsequent chemical transformations.18 Fluoropyridinium 8 proved to be the optimal reagent to yield β-fluorinated amine 9 (75%), and N-chlorosuccinimide enabled effective chlorination to provide β-chlorinated piperidine 10 (85%). These reductive halogenation reactions could be modified by replacing the borane reductant with a Grignard reagent, enabling the formation of a C–C bond in the α-position of the piperidines. Vinyl and p-tolyl Grignard reagents were used in both fluorination and chlorination procedures, providing trifunctionalized piperidines 11a–d in high yields (63–81%). Only a single diastereomer was observed to form under these reaction conditions, suggesting an ordered transition state controlling the approach of the nucleophile to the iminium intermediate (12). Desulfonylation of 11b could be achieved using magnesium in methanol,19 to afford stereodefined β-fluoropiperidine 13 with good selectivity. Combining initial chlorination with a hydroxide trap led to ring-opening of the piperidine and formation of formamide 14. Arylation β to the amine was achieved using an aryliodonium salt in the presence of a copper catalyst,20 producing amine 15 in 22% yield. The formation of a congested quaternary center likely contributes to this low yield. Finally, reaction of 2a with a diazonium salt induced ring-opening and loss of a methylene unit, producing hydrazone 16 in 52% yield via a Japp–Klingemann reaction.21
Conclusions
In conclusion, we have developed a straightforward process for the β-C–H functionalization of piperidines. The reactions combine piperidines and sodium sulfinates, under the action of NIS, to provide enaminyl sulfone products. The process is achieved under mild conditions, and shows good functional group tolerance. We also establish that the resultant cyclic enaminyl sulfones are versatile templates for further elaboration. We envisage that this approach will expedite the generation of diverse compound libraries for use in drug discovery.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by GSK via the GSK/University of Strathclyde Centre for Doctoral Training in Synthetic and Medicinal Chemistry. We also thank Sean Lynn and Richard Upton (both GSK) for their assistance with NMR analysis.References
- (a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (b) P. A. Petukhov, J. Zhang, C. Z. Wang, Y. P. Ye, K. M. Johnson and A. P. Kozikowski, J. Med. Chem., 2004, 47, 3009–3018 CrossRef CAS PubMed; (c) M. Nakanishi, C. Tashiro, T. Munakata, K. Araki, T. Tsumagari and H. Imamura, J. Med. Chem., 1970, 13, 644–648 CrossRef CAS PubMed; (d) B. Pati and S. Banerjee, J. Pharma Res., 2012, 5, 5493–5509 CAS.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- (a) J. E. Spangler, Y. Kobayashi, P. Verma, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 11876–11879 CrossRef CAS PubMed; (b) S. J. Pastine, D. V. Gribkov and D. Sames, J. Am. Chem. Soc., 2006, 128, 14220–14221 CrossRef CAS PubMed; (c) J. He, L. G. Hamann, H. M. L. Davies and R. E. J. Beckwith, Nat. Commun., 2015, 6, 5943–5951 CrossRef PubMed; (d) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC; (e) E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel and B. U. W. Maes, Chem.–Eur. J., 2012, 18, 10092–10142 CrossRef CAS PubMed.
- (a) J. J. Topczewski, P. J. Cabrera, N. I. Saper and M. S. Sanford, Nature, 2016, 531, 220–224 CrossRef CAS PubMed; (b) D. P. Affron, O. A. Davis and J. A. Bull, Org. Lett., 2014, 16, 4956–4959 CrossRef CAS PubMed; (c) G. Asensio, M. E. Gonzalez-Nunez, C. B. Bernardini, R. Mello and W. Adam, J. Am. Chem. Soc., 1993, 115, 7250–7253 CrossRef CAS; (d) M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 12796–12799 CrossRef CAS PubMed; (e) N. Takasu, K. Oisaki and M. Kanai, Org. Lett., 2013, 15, 1918–1921 CrossRef CAS PubMed; (f) W. Chen, Y. Kang, R. G. Wilde and D. Seidel, Angew. Chem., Int. Ed., 2014, 53, 5179–5182 CAS; (g) L. Ma, A. Paul, M. Breugst and D. Seidel, Chem.–Eur. J., 2016, 22, 18179–18189 CrossRef CAS PubMed; (h) J. M. Howell, K. Feng, J. R. Clark, L. J. Trzepkowski and M. C. White, J. Am. Chem. Soc., 2015, 137, 14590–14593 CrossRef CAS PubMed; (i) D. M. Schultz, F. Lévesque, D. A. DiRocco, M. Reibarkh, Y. Ji, L. A. Joyce, J. F. Dropinski, H. Sheng, B. D. Sherry and I. W. Davies, Angew. Chem., Int. Ed., 2017, 56, 15274–15278 CrossRef CAS PubMed.
- (a) E. Fromm and J. Wittmann, Ber. Dtsch. Chem. Ges., 1908, 41, 2264–2273 CrossRef CAS; (b) H. Tucker, J. W. Crook and G. J. Chesterson, J. Med. Chem., 1988, 31, 954–959 CrossRef CAS PubMed; (c) M. Couderchet, J. Schmalfuß and P. Böger, Pestic. Sci., 1998, 52, 381–387 CrossRef CAS; (d) Smithkline Beecham Corporation, J. Busch-Petersen and Glaxosmithkline Llc, Ca. Pat., CA2650009 C, 2014; (e) Vertex Pharmaceuticals Incorporated, US Pat., US2013/115310, 2013; (f) Bristol-Myers Squibb Company, WO2015/103510, 2015.
- (a) N. Simpkins, Sulfones in Organic Synthesis, Pergamon Press, Oxford, 1st edn, 1993 Search PubMed; (b) N. S. Simpkins, Tetrahedron, 1990, 46, 6951–6984 CrossRef CAS.
- (a) L. K. Liu, Y. Chi and K.-Y. Jen, J. Org. Chem., 1980, 45, 406–410 CrossRef CAS; (b) H. Goldwhite, M. S. Gibson and C. Harris, Tetrahedron, 1964, 20, 1613–1624 CrossRef CAS; (c) T. G. Back and S. Collins, J. Org. Chem., 1981, 46, 3249–3256 CrossRef CAS; (d) Y. H. Kang and J. L. Kice, J. Org. Chem., 1984, 49, 1507–1511 CrossRef CAS; (e) D. Duan and X. Huang, Synlett, 1999, 1999, 317–318 CrossRef; (f) X. Li, X. Shi, M. Fang and X. Xu, J. Org. Chem., 2013, 78, 9499–9504 CrossRef CAS PubMed; (g) Y. Xi, B. Dong, E. J. McClain, Q. Wang, T. L. Gregg, N. G. Akhmedov, J. L. Petersen and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 4657–4661 CrossRef CAS PubMed; (h) Y. Xu, J. Zhao, X. Tang, W. Wu and H. Jiang, Adv. Synth. Catal., 2014, 356, 2029–2039 CrossRef CAS; (i) N. Taniguchi, Tetrahedron, 2014, 70, 1984–1990 CrossRef CAS.
- (a) B. P. Bandgar, S. V. Bettigeri and J. Phopase, Org. Lett., 2004, 6, 2105–2108 CrossRef CAS PubMed; (b) S. Cacchi, G. Fabrizi, A. Goggiamani, L. M. Parisi and R. Bernini, J. Org. Chem., 2004, 69, 5608–5614 CrossRef CAS PubMed; (c) A. U. Meyer, S. Jäger, D. Prasad Hari and B. König, Adv. Synth. Catal., 2015, 357, 2050–2054 CrossRef CAS; (d) P. Katrun, S. Chiampanichayakul, K. Korworapan, M. Pohmakotr, V. Reutrakul, T. Jaipetch and C. Kuhakarn, Eur. J. Org. Chem., 2010, 5633–5641 CrossRef CAS; (e) Y. Sun, A. Abdukader, D. Lu, H. Zhang and C. Liu, Green Chem., 2017, 19, 1255–1258 RSC.
- (a) E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2014, 53, 10204–10208 CrossRef CAS PubMed; (b) A. S. Deeming, C. J. Russell and M. C. Willis, Angew. Chem., Int. Ed., 2016, 55, 747–750 CrossRef CAS PubMed; (c) Y. Chen and M. C. Willis, Chem. Sci., 2017, 8, 3249–3253 RSC; (d) E. J. Emmett and M. C. Willis, Asian J. Org. Chem., 2015, 4, 602–611 CrossRef CAS; (e) A. S. Deeming and M. C. Willis, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, Chichester, UK, 2016, pp. 1–4 Search PubMed.
- R. J. Griffiths, G. A. Burley and E. P. A. Talbot, Org. Lett., 2017, 19, 870–873 CrossRef CAS PubMed.
- J. Lai, L. Chang and G. Yuan, Org. Lett., 2016, 18, 3194–3197 CrossRef CAS PubMed.
- (a) M. Chen, Z.-T. Huang and Q.-Y. Zheng, Org. Biomol. Chem., 2014, 12, 9337–9340 RSC; (b) Y. Cai, R. Zhang, D. Sun, S. Xu and Q. Zhou, Synlett, 2017, 28, 1630–1635 CrossRef CAS.
- L. K. Liu, Y. Chi and K.-Y. Jen, J. Org. Chem., 1980, 45, 406–410 CrossRef CAS.
- (a) S. W. Wright and K. N. Hallstrom, J. Org. Chem., 2006, 71, 1080–1084 CrossRef CAS PubMed; (b) A. García-Rubia, B. Urones, R. Gómez Arrayás and J. C. Carretero, Angew. Chem., Int. Ed., 2011, 50, 10927–10931 CrossRef PubMed; (c) E. Wedekind and D. Schenk, Ber. Dtsch. Chem. Ges., 1911, 44, 198–202 CrossRef CAS; (d) J. F. King, Acc. Chem. Res., 1975, 8, 10–17 CrossRef CAS.
- (a) W. Zhu, G. Cai and D. Ma, Org. Lett., 2005, 7, 5545–5548 CrossRef CAS PubMed; (b) D.-J. Zhang, M.-S. Xie, G.-R. Qu, Y.-W. Gao and H.-M. Guo, Org. Lett., 2016, 18, 820–823 CrossRef CAS PubMed.
- A. E. Lanzilotti, R. Littell, W. J. Fanshawe, T. C. McKenzie and F. M. Lovell, J. Org. Chem., 1979, 44, 4809–4813 CrossRef CAS.
- (a) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed; (b) C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061–5084 CrossRef CAS PubMed.
- (a) M. Dryzhakov, E. Richmond, G. Li and J. Moran, J. Fluorine Chem., 2017, 193, 45–51 CrossRef CAS; (b) M. Dryzhakov and J. Moran, ACS Catal., 2016, 6, 3670–3673 CrossRef CAS; (c) F. González-Bobes and G. C. Fu, J. Am. Chem. Soc., 2006, 128, 5360–5361 CrossRef PubMed; (d) H. Tateno, Y. Matsumura, K. Nakabayashi, H. Senboku and M. Atobe, RSC Adv., 2015, 5, 98721–98723 RSC; (e) M. Sidera and S. P. Fletcher, Nat. Chem., 2015, 7, 935–939 CrossRef CAS PubMed.
- T. Shibue and Y. Fukuda, J. Org. Chem., 2014, 79, 7226–7231 CrossRef CAS PubMed.
- A. Bigot, A. E. Williamson and M. J. Gaunt, J. Am. Chem. Soc., 2011, 133, 13778–13781 CrossRef CAS PubMed.
- T. Laue and A. Plagens, Named Organic Reactions, Wiley, 2nd edn, 2005 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04900e |
| ‡ Present address: Pharmaron, Hertford Rd, Hoddesdon, EN11 9BU, UK. E-mail: E-mail: eric.talbot@pharmaron-uk.com |
| This journal is © The Royal Society of Chemistry 2018 |