
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNi-catalysed regioselective 1,2-diarylation of unactivated olefins by stabilizing Heck intermediates as pyridylsilyl-coordinated transient metallacycles†
Surendra
Thapa‡
,
Roshan K.
Dhungana‡
,
Rajani Thapa
Magar
,
Bijay
Shrestha
,
Shekhar
KC
and
Ramesh
Giri
 *
*
Department of Chemistry & Chemical Biology, The University of New Mexico, Albuquerque, NM 87131, USA. E-mail: rgiri@unm.edu
First published on 27th November 2017
Abstract
We report a Ni-catalysed diarylation of unactivated olefins in dimethylpyridylvinylsilane by intercepting Heck C(sp3)–NiX intermediates, derived from aryl halides, with arylzinc reagents. This approach utilizes a modifiable pyridylsilyl moiety as a coordinating group that plays a dual role of intercepting oxidative addition species to promote Heck carbometallation, and stabilizing the Heck C(sp3)–NiX intermediates as transient metallacycles to suppress β-hydride elimination, and facilitate transmetalation/reductive elimination. This method affords 1,2-diarylethylsilanes, which can be readily oxidized to 1,2-diarylethanols that occur as structural motifs in 3-aryl-3,4-dihydroisocoumarin and dihydrostilbenoid natural products.
Introduction
Interception of Heck C(sp3)–[M] intermediates with carbon nucleophiles by cross-coupling is an attractive approach to construct simultaneously two carbon–carbon (C–C) bonds across an olefin in one synthetic step.1 This olefin dicarbofunctionalization process integrates the Heck reaction and cross-coupling in one synthetic platform (Scheme 1, path A) to rapidly build molecular complexity, an endeavour that would otherwise require a multi-step process if pursued through traditional synthetic disconnection strategies. However, execution of such a process on an unactivated olefin remains formidably challenging especially with transition metals such as Pd. The difficulty arises due to the sheer requirement to overcome two of the most fundamental Pd-catalysed processes as side reactions – direct cross-coupling between an organohalide and an organometallic reagent prior to olefin insertion (Scheme 1, path B), and the Heck reaction by β-hydride (β-H) elimination from the C(sp3)–[M] intermediates formed after carbometallation to an olefin (Scheme 1, path C).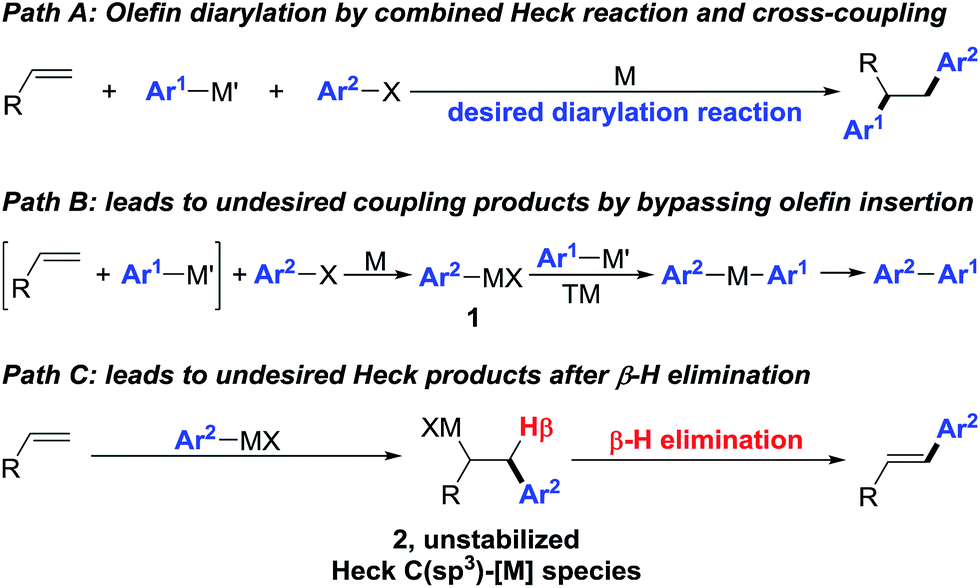 | ||
| Scheme 1 Two well-known transformations working as side reactions during regioselective olefin diarylation. | ||
Prior reports have exploited two strategies to overcome these problems and enable the transition metal-catalysed three-component olefin dicarbofunctionalization reactions – (1) the use of dienes and styrenes as substrates that would stabilize the Heck C(sp3)–[M] intermediates by intrinsic π-allyl and π-benzyl formation,2 and (2) the generation and addition of alkyl radicals to olefins.3 In simple olefins that lack the means to stabilize the Heck C(sp3)–[M] intermediates,4 reactions generally follow the Heck carbometallation/β-H elimination/M-H reinsertion cascade to furnish 1,1-dicarbofunctionalized products.5
Recently, we6 developed a (cod)2Ni-catalysed three-component dicarbofuntionalization of olefins in which we strategically employed imines as a readily removable coordinating group to facilitate the reaction.7 The idea was to intercept the organonickel species 3 by a bidentate coordination mode, promote intramolecular Heck carbonickellation onto the bound olefin and stabilize the resultant Heck C(sp3)–[Ni] intermediate 6 as a transient metallacycle (Scheme 2). We envisioned that this strategy would afford sufficient residence time for the Heck C(sp3)–[Ni] species 6 in order to promote the requisite transmetalation/reductive elimination steps, and furnish the expected product. We believe that this approach should operate regardless of the reaction proceeding via a Ni0/NiII or NiI/NiIII catalytic cycle. Herein, we report diarylation of unactivated olefins in pyridylvinylsilanes that relies upon the same strategy of stabilizing the Heck C(sp3)–[Ni] intermediates as transient five-membered metallacycles by pyridylsilyl coordination.8
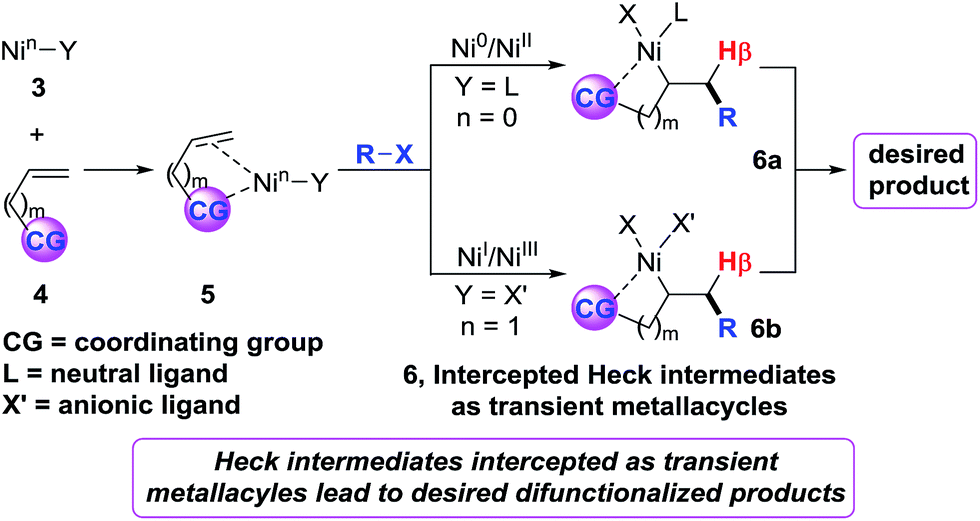 | ||
| Scheme 2 Strategy for olefin dicarbofunctionalization. | ||
Results and discussion
Towards our long-term goal of olefin dicarbofunctionalization by cross-coupling,4a,k we aspired to expand the scope of our three-component diarylation of olefins to silicon-based molecules. We believe that development of such a method could provide rapid access to complex differently substituted 1,2-diarylethylsilanes, which are difficult to synthesize.9 The 1,2-diarylethylsilanes could then be readily oxidized to 1,2-diarylethanols that occur as structural motifs in wide range of 3-aryl-3,4-dihydroisocoumarin and dihydrostilbenoid natural products.10 In this process, we examined the diarylation of pyridylvinylsilane 7 with 4-bromobenzotrifluoride and PhZnI (Table 1). Examination of various parameters revealed that the reaction furnished the expected product 15a in best yield (79%) using 2 mol% NiBr2 as a catalyst in NMP at room temperature (entry 1). We then investigated substituted pyridyl (8 and 10), 8-quinolinyl (9), dipyridyl (11), N,N-dimethylanilinyl (12), o-anisolyl (13) and trimethoxysilyl (14) as coordinating groups where the nitrogen or oxygen atoms could coordinate to Heck C(sp3)–[Ni] intermediates to generate 4-, 5- or 6-membered transient metallacycles (entries 2–8). Among them, only the 4-methylpyridyl (8) and 8-quinolinyl (9) afforded the expected products 15b–c in good yields (entries 2–3).11 Running the reaction for a shorter time (15 h) formed the product in slightly lower yield (entry 9). Other Ni-catalysts such as Ni(cod)2 and Ni(PPh3)4 also generated the product in lower yields (entries 10 and 11). The lower yields with Ni(cod)2 and Ni(PPh3)4 could be rationalized based on difficulty to displace strongly coordinating ligands such as 1,5-cyclooctadiene (cod) and Ph3P by the substrate imine or olefin. Using DMF or DMA as a solvent furnished product 15a in moderate yields (entry 12). Other solvents such as DMSO, dioxane, MeCN, benzene and THF formed the product 15a in <15% yield (entries 13 and 14). The use of a Ni-catalyst was found to be critical for this transformation as other catalysts based on Pd, Co, Fe and Cu did not catalyse the reaction (entries 15 and 16).|
|
||||
|---|---|---|---|---|
| Entry | Reaction condition deviation | Vinylsilanes | Product | Yields of 15 (%) |
| a Yields were determined by 1H NMR using pyrene as an internal standard. Value in parenthesis is the isolated yield from a 0.5 mmol scale reaction. b Heck product was formed in >90% NMR yield with Co(OAc)2. | ||||
| 1 | None | 7 | 15a | 79 (72) |
| 2 | None | 8 | 15b | 51 |
| 3 | None | 9 | 15c | 56 |
| 4 | None | 10 | 15d | 0 |
| 5 | None | 11 | 15e | 0 |
| 6 | None | 12 | 15f | 0 |
| 7 | None | 13 | 15g | 0 |
| 8 | None | 14 | 15h | 0 |
| 9 | 15 h | 7 | 15a | 62 |
| 10 | (cod)2Ni instead of NiBr2 | 7 | 15a | 35 |
| 11 | (Ph3P)4Ni instead of NiBr2 | 7 | 15a | 30 |
| 12 | DMF or DMA instead of NMP | 7 | 15a | 40–44 |
| 13 | DMSO, dioxane or MeCN instead of NMP | 7 | 15a | 10–15 |
| 14 | Benzene of THF instead of NMP | 7 | 15a | <5 |
| 15 | Cul or FeCl3 instead of NiBr2 | 7 | 15a | 0 |
| 16 | Pd(OAc)2 or Co(OAc)2 instead of NiBr2 | 7 | 15a | 0b |

We note that the reaction does not form products arising from the Hiyama coupling12 despite the fact pyridylvinylsilanes are known to undergo such a reaction.13 The current reaction also proceeds with absolute regioselectivity with terminal arylation during Heck carbometallation, a result that is in sharp contrast to the undirected Ni-catalysed Heck reactions that proceed with excellent selectivity for internal arylation.14 The regioselectivity of the products was confirmed by oxidation of 1,2-diarylethylsilanes 19 and 23 (Table 2) to the corresponding known 1,2-diarylethanols 45 and 46 (vide infra, Scheme 3), respectively.15 We hypothesize that the remarkable terminal selectivity for Heck carbometallation in our reaction could be a function of both the coordinating effect of the pyridyl group that strives for forming stable and tight five-membered metallacycles after carbometallation on the vinyl group, and the β-cation stabilizing effect of Si due to hyperconjugation.16 Prior work on Pd- and Fe-catalysed, directed Heck reaction of dimethyl(2-pyridyl)vinylsilane has also shown that the reaction proceeds with high regioselectivity for terminal arylation.17
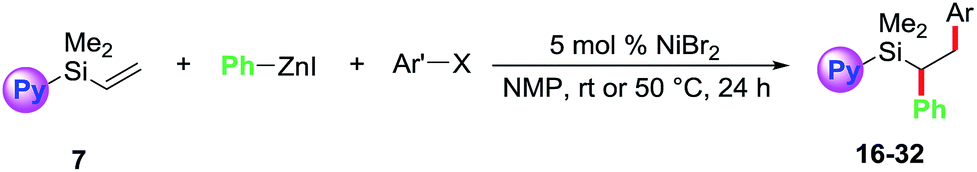
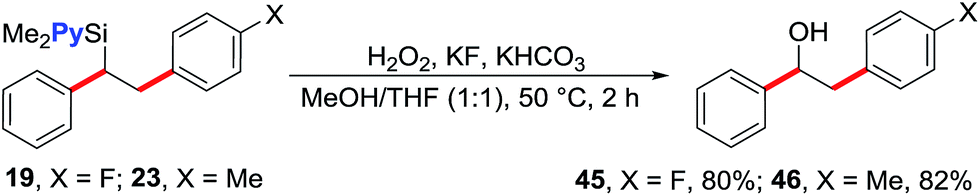 | ||
| Scheme 3 Transformation of silyl group to alcohol. | ||
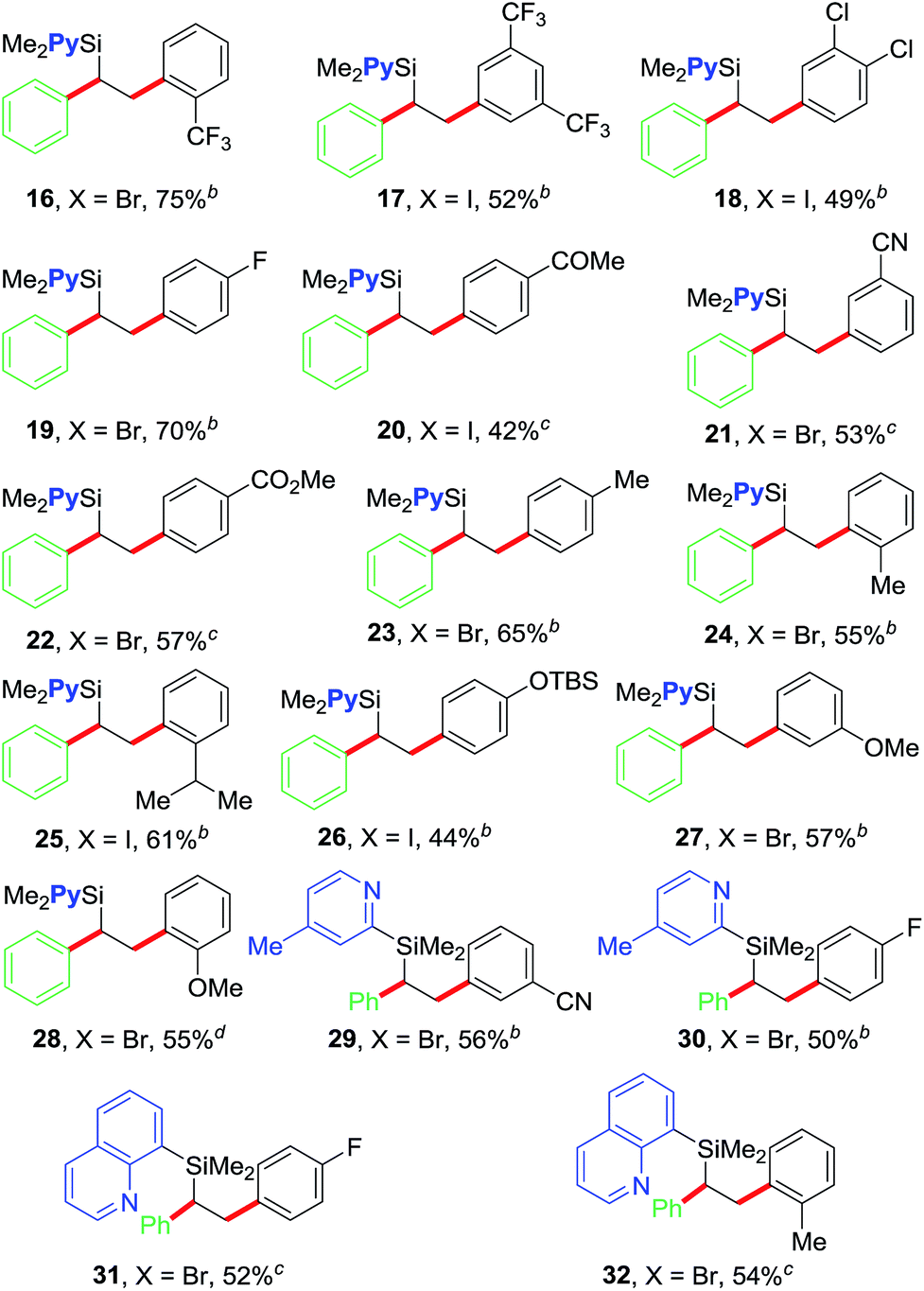
After optimizing the reaction conditions, we examined the scope of the current olefin diarylation reaction (Table 2). In general, reactions of electron-deficient aryl halides reacted at room temperature (20–22, 29 and 31) and those with electron-rich substituents required 50 °C (23–28 and 30). In addition, reactions typically required 2–5 mol% NiBr2. A wide variety of electron-rich, neutral and electron-deficient aryl iodides can be utilized as coupling partners along with the pyridylvinylsilane 7, which furnishes variously substituted 1,2-diarylethylsilanes 16–28 in good yields.18 The reaction tolerates a variety of functional groups such as Me, OMe, OTBS, CF3, F, Cl, COMe, CO2Me and CN. The reaction also works well with aryl halides containing ortho-substituents (16, 24, 28 and 32) including sterically hindered isopropyl group (25).
We further examined the scope of the reaction with respect to arylzinc reagents (Table 3). Electron-rich, deficient and neutral arylzinc reagents containing Me, OMe, F and CF3 can be used as coupling partners19 along with aryl halides bearing various functional groups like Me, OMe, CF3, Cl, COMe, CO2Me and CN, which afford the 1,2-diarylated products in good yields (33–44). The reaction also tolerates ortho-substituted arylzinc reagents (43 and 44).
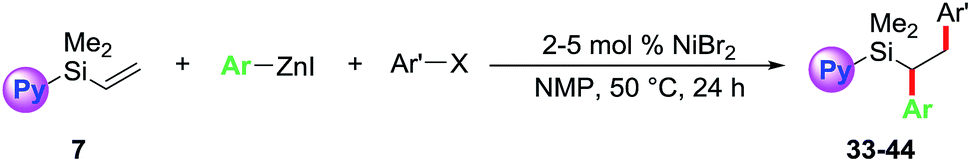
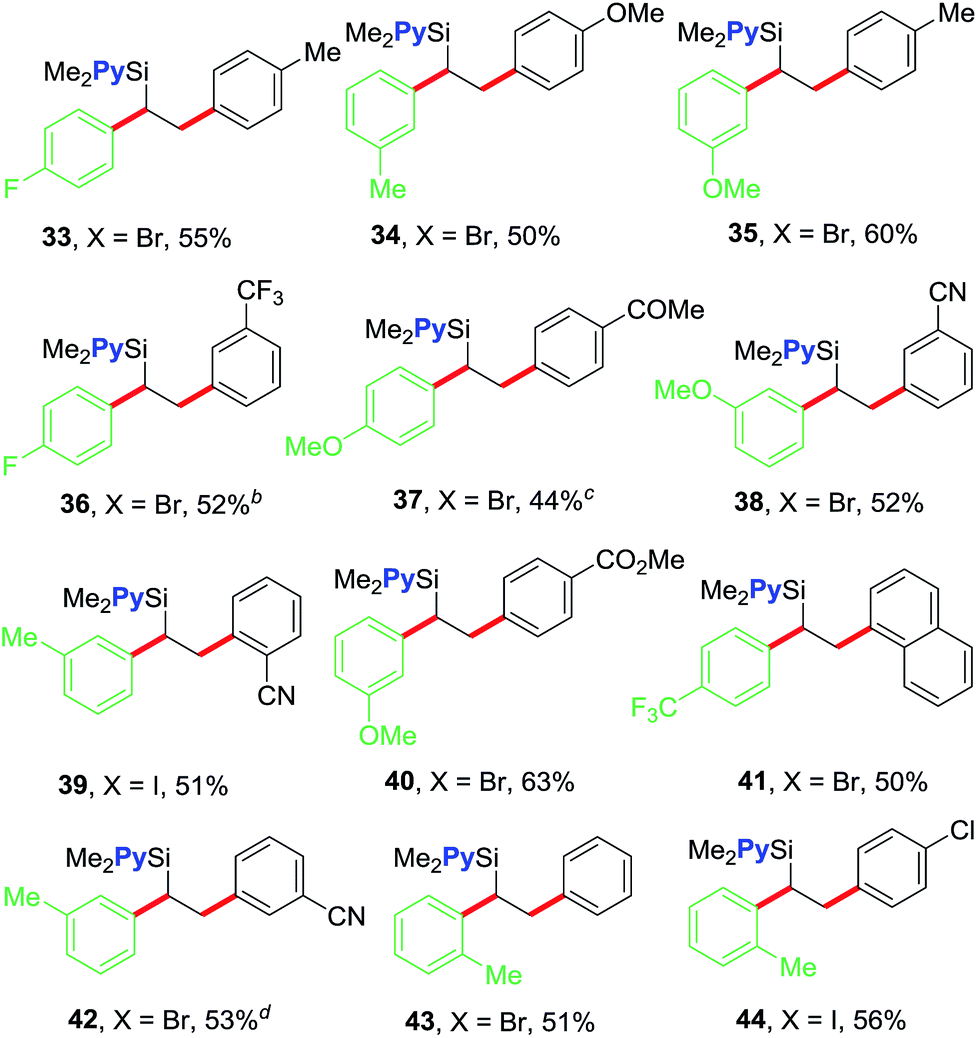
The 1,2-diarylethylsilane products can be readily transformed into 1,2-diarylethanol by oxidation (Scheme 3). For example, we show that the products 19 and 23 can be efficiently oxidized to 2-(4-fluorophenyl)-1-phenylethanol (45) and 1-phenyl-2-(p-tolyl)ethanol (46), respectively, in excellent yields by reacting them with H2O2 in the presence of KF and KHCO3.20
We conducted further studies in order to probe the role of the pyridyl group (Scheme 4). We performed the reaction of phenylvinylsilane 47 with 4-bromobenzotrifluoride and PhZnI under the standard conditions. Despite Ni being a good catalyst for the Heck reaction,14,21 phenylvinylsilane 47 did not afford any Heck or the dicarbofunctionalized product. Only the direct cross-coupling product was formed in 81% yield. This result indicates that the pyridyl group in pyridylvinylsilane 7 is indeed required for both the Heck carbometallation of Ar[Ni] on the vinyl group, and stabilizing the Heck C(sp3)–[Ni] intermediates as five-membered metallacycles prior to the delivery of the desired 1,2-diarylated products via transmetalation/reductive elimination processes.
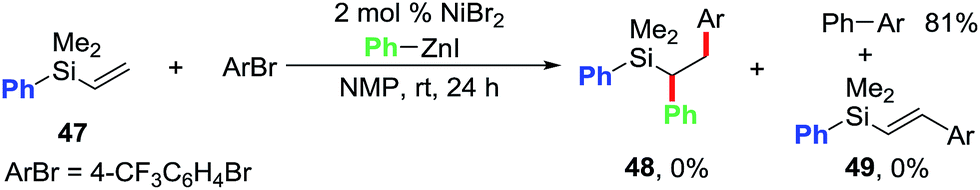 | ||
| Scheme 4 Establishing the role of pyridylsilyl group. | ||
We also conducted a radical clock experiment in order to determine if aryl radicals were formed during the reaction of aryl halides with the Ni-catalyst (Scheme 5). For this purpose, we utilized 1-(but-3-enyl)-2-iodobenzene (50) as a radical probe. The aryl radical 52 generated from the radical probe 50 is known to undergo a fast radical cyclization with a kobs of 5.0 × 108 s−1 to 1-methyldihydroindenyl radical 53.22 When we reacted the radical clock 50 with the pyridylvinylsilane 7 and PhZnI using 5 mol% NiBr2 as a catalyst, the diarylated product 51 was formed in 75% isolated yield. The cyclized products 54 and 55, expected to arise if the aryl radical 52 was generated, were not observed. This experiment clearly indicates that oxidative addition of ArX to the Ni-catalyst proceeds without the generation of aryl free radicals.
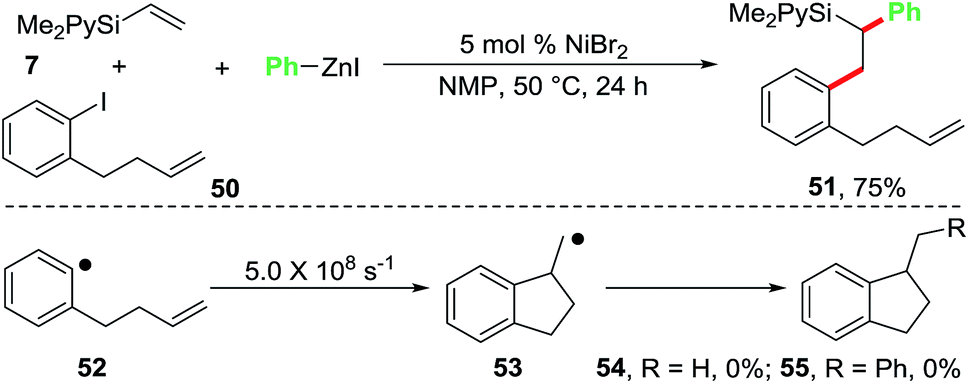 | ||
| Scheme 5 Radical clock experiment. | ||
Based on the control and radical clock experiments, and the observation of complete regioselectivity, we propose that the current diarylation reaction proceeds via a Ni(0)/Ni(II) catalytic cycle as outlined in Scheme 6a. Herein, the reaction proceeds via initial oxidative addition of ArX to Ni(0) bound to pyridylvinylsilane 7 followed by migratory insertion, transmetalation and reductive elimination steps to furnish the 1,2-diarylated products. An alternative non-radical Ni(I)/Ni(III) catalytic cycle as outlined in Scheme 6b proceeding via initial transmetalation of PhZnI23 followed by either oxidative addition/reductive elimination or migratory insertion/oxidative addition/reductive elimination sequence can be readily discounted because such a process would lead to products where the regioselectivity would be either lost or opposite to the one observed experimentally.
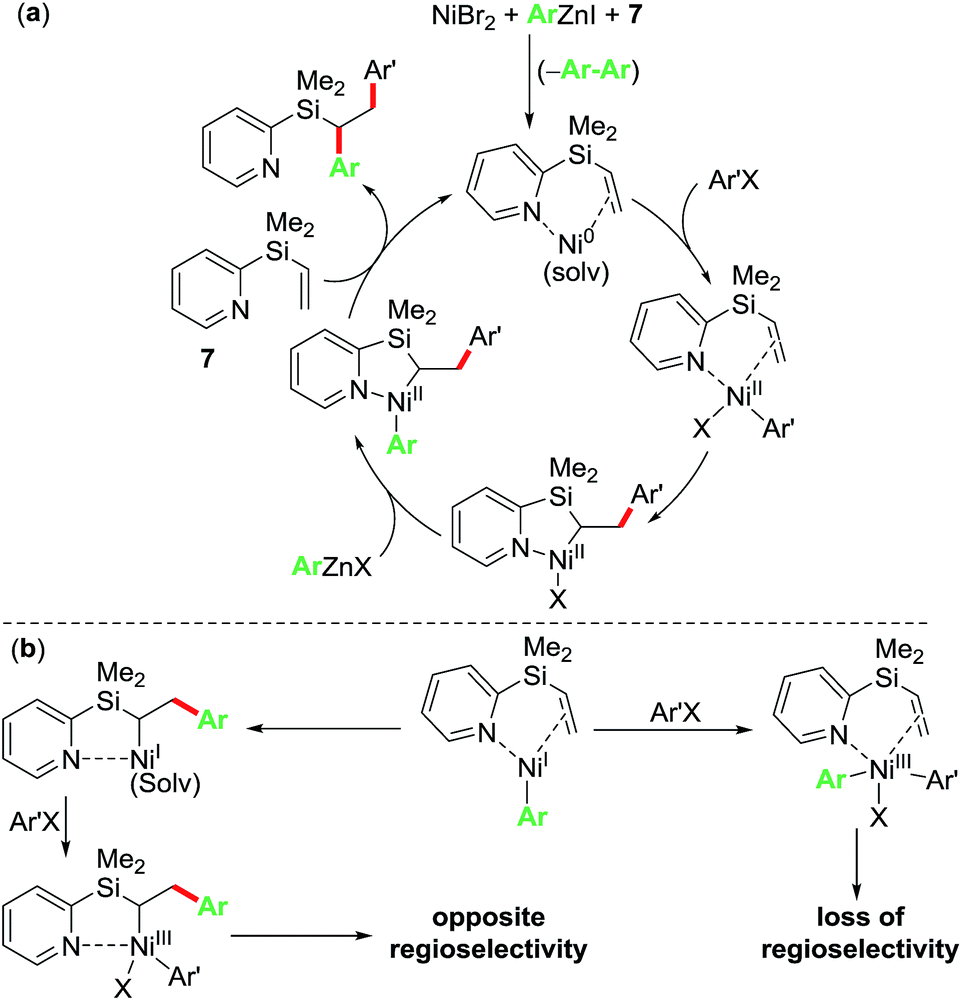 | ||
| Scheme 6 Proposed catalytic cycle. | ||
Conclusions
In summary, we have developed a Ni-catalysed diarylation of unactivated olefins in pyridylvinylsilanes by cross-coupling using aryl halides and arylzinc reagents as carbon sources. The reaction affords 1,2-diarylethylsilanes, which can be readily converted to 1,2-diarylethanols that are structural motifs in natural products and biologically important molecules. The reaction also tolerates various functional groups that are synthetically important. Preliminary studies indicate that the reaction proceeds via a Ni(0)/Ni(II) catalytic cycle.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the University of New Mexico (UNM) and the National Science Foundation (NSF CHE-1554299) for financial support, and upgrades to the NMR (NSF grants CHE08-40523 and CHE09-46690) and MS Facilities.Notes and references
- For dicarbofunctionalization of activated olefins by conjugate addition/enolate interception, see: (a) H.-C. Guo and J.-A. Ma, Angew. Chem., Int. Ed., 2006, 45, 354 CrossRef CAS PubMed; (b) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801 CrossRef CAS PubMed.
- (a) L. Liao, R. Jana, K. B. Urkalan and M. S. Sigman, J. Am. Chem. Soc., 2011, 133, 5784 CrossRef CAS PubMed; (b) X. Wu, H.-C. Lin, M.-L. Li, L.-L. Li, Z.-Y. Han and L.-Z. Gong, J. Am. Chem. Soc., 2015, 137, 13476 CrossRef CAS PubMed; (c) Z. Kuang, K. Yang and Q. Song, Org. Lett., 2017, 19, 2702 CrossRef CAS PubMed; (d) J. Terao, S. Nii, F. A. Chowdhury, A. Nakamura and N. Kambe, Adv. Synth. Catal., 2004, 346, 905 CrossRef CAS; (e) K. Mizutani, H. Shinokubo and K. Oshima, Org. Lett., 2003, 5, 3959 CrossRef CAS PubMed.
- (a) A. García-Domínguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835 CrossRef PubMed; (b) J.-W. Gu, Q.-Q. Min, L.-C. Yu and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 12270 CrossRef CAS PubMed; (c) B. J. Stokes, L. Liao, A. M. de Andrade, Q. Wang and M. S. Sigman, Org. Lett., 2014, 16, 4666 CrossRef CAS PubMed.
- For examples of two-component dicarbofunctionalizations of tethered olefins, see: (a) R. K. Dhungana, B. Shrestha, R. Thapa-Magar, P. Basnet and R. Giri, Org. Lett., 2017, 19, 2154 CrossRef CAS PubMed; (b) G. Balme, D. Bouyssi, T. Lomberget and N. Monteiro, Synthesis, 2003, 2003, 2115 CrossRef; (c) G. Fournet, G. Balme and J. Gore, Tetrahedron Lett., 1987, 28, 4533 CrossRef CAS; (d) J. G. Kim, Y. H. Son, J. W. Seo and E. J. Kang, Eur. J. Org. Chem., 2015, 2015, 1781 CrossRef CAS; (e) V. B. Phapale, E. Buñuel, M. García-Iglesias and D. J. Cárdenas, Angew. Chem., Int. Ed., 2007, 46, 8790 CrossRef CAS PubMed; (f) M. Nakamura, S. Ito, K. Matsuo and E. Nakamura, Synlett, 2005, 2005, 1794 CrossRef; (g) K. Wakabayashi, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2001, 123, 5374 CrossRef CAS PubMed; (h) C.-S. Yan, Y. Peng, X.-B. Xu and Y.-W. Wang, Chem.–Eur. J., 2012, 18, 6039 CrossRef CAS PubMed; (i) B. Seashore-Ludlow and P. Somfai, Org. Lett., 2012, 14, 3858 CrossRef CAS PubMed; (j) R. Grigg, J. Sansano, V. Santhakumar, V. Sridharan, R. Thangavelanthum, M. Thornton-Pett and D. Wilson, Tetrahedron, 1997, 53, 11803 CrossRef CAS; (k) S. Thapa, P. Basnet and R. Giri, J. Am. Chem. Soc., 2017, 139, 5700 CrossRef CAS PubMed; (l) W. You and M. K. Brown, J. Am. Chem. Soc., 2015, 137, 14578 CrossRef CAS PubMed; (m) W. You and M. K. Brown, J. Am. Chem. Soc., 2014, 136, 14730 CrossRef CAS PubMed; (n) H. Cong and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 3788 CrossRef CAS PubMed; (o) C. M. McMahon, M. S. Renn and E. J. Alexanian, Org. Lett., 2016, 18, 4148 CrossRef CAS PubMed; (p) A. Vaupel and P. Knochel, J. Org. Chem., 1996, 61, 5743 CrossRef CAS; (q) T. Ishiyama, M. Murata, A. Suzuki and N. Miyaura, J. Chem. Soc., Chem. Commun., 1995, 295 RSC; (r) J. A. Walker, K. L. Vickerman, J. N. Humke and L. M. Stanley, J. Am. Chem. Soc., 2017, 139, 10228 CrossRef CAS PubMed.
- (a) V. Saini and M. S. Sigman, J. Am. Chem. Soc., 2012, 134, 11372 CrossRef CAS PubMed; (b) E. W. Werner, K. B. Urkalan and M. S. Sigman, Org. Lett., 2010, 12, 2848 CrossRef CAS PubMed; (c) V. Saini, L. Liao, Q. Wang, R. Jana and M. S. Sigman, Org. Lett., 2013, 15, 5008 CrossRef CAS PubMed; (d) K. B. Urkalan and M. S. Sigman, Angew. Chem., Int. Ed., 2009, 48, 3146 CrossRef CAS PubMed.
- B. Shrestha, P. Basnet, R. K. Dhungana, S. Kc, S. Thapa, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2017, 139, 10653 CrossRef CAS PubMed.
- For other examples of directed olefin difunctionalization, see: with Pd: (a) S. Yahiaoui, A. Fardost, A. Trejos and M. Larhed, J. Org. Chem., 2011, 76, 2433 CrossRef CAS PubMed; (b) E. P. A. Talbot, T. d. A. Fernandes, J. M. McKenna and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 4101 CrossRef CAS PubMed; (c) S. R. Neufeldt and M. S. Sanford, Org. Lett., 2013, 15, 46 CrossRef CAS PubMed; (d) Z. Liu, T. Zeng, K. S. Yang and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 15122 CrossRef CAS PubMed; (e) J. Derosa, V. T. Tran, M. N. Boulous, J. S. Chen and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 10657 CrossRef CAS PubMed.
- For the use of pyridylsilyl as a directing group for C–H activation, see: (a) A. V. Gulevich, F. S. Melkonyan, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2012, 134, 5528 CrossRef CAS PubMed; (b) N. Chernyak, A. S. Dudnik, C. Huang and V. Gevorgyan, J. Am. Chem. Soc., 2010, 132, 8270 CrossRef CAS PubMed; for a review on the use of pyridylsilyl as a directing group, see: (c) K. Itami and J.-i. Yoshida, Synlett, 2006, 2006, 157 CrossRef.
- For prior reports each showing the synthesis of one 1,2-diarylethylsilane, see: (a) C. K. Hazra, N. Gandhamsetty, S. Park and S. Chang, Nat. Commun., 2016, 7, 13431 CrossRef CAS PubMed; (b) A. Saxena and H. W. Lam, Chem. Sci., 2011, 2, 2326 RSC; (c) S. Nii, J. Terao and N. Kambe, J. Org. Chem., 2004, 69, 573 CrossRef CAS PubMed.
- (a) J. Chen, L. Zhou, C. K. Tan and Y.-Y. Yeung, J. Org. Chem., 2012, 77, 999 CrossRef CAS PubMed; (b) G. C. Tron, T. Pirali, G. Sorba, F. Pagliai, S. Busacca and A. A. Genazzani, J. Med. Chem., 2006, 49, 3033 CrossRef CAS PubMed; (c) J. A. Baur and D. A. Sinclair, Nat. Rev. Drug Discovery, 2006, 5, 493 CrossRef CAS PubMed; (d) A. Cirla and J. Mann, Nat. Prod. Rep., 2003, 20, 558 RSC; (e) S. B. Singh and G. R. Pettit, Synth. Commun., 1987, 17, 877 CrossRef CAS.
- Since 2-OMe group is known to decrease the basicity of the pyridyl nitrogen, it could weaken pyridyl N-binding to Ni resulting in no reactivity of the olefin 10. See: R. A. Murphy and R. Sarpong, Org. Lett., 2012, 14, 632 CrossRef CAS PubMed . As such, 23% of direct cross-coupling product was formed in this reaction.
- (a) D. A. Powell and G. C. Fu, J. Am. Chem. Soc., 2004, 126, 7788 CrossRef CAS PubMed; (b) X. Dai, N. A. Strotman and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 3302 CrossRef CAS PubMed.
- K. Itami, T. Nokami and J.-i. Yoshida, J. Am. Chem. Soc., 2001, 123, 5600 CrossRef CAS PubMed.
- (a) R. Matsubara, A. C. Gutierrez and T. F. Jamison, J. Am. Chem. Soc., 2011, 133, 19020 CrossRef CAS PubMed; (b) E. A. Standley and T. F. Jamison, J. Am. Chem. Soc., 2013, 135, 1585 CrossRef CAS PubMed; (c) S. Z. Tasker, A. C. Gutierrez and T. F. Jamison, Angew. Chem., Int. Ed., 2014, 53, 1858 CrossRef CAS PubMed.
- D. K. Nielsen and A. G. Doyle, Angew. Chem., Int. Ed., 2011, 50, 6056 CrossRef CAS PubMed.
- S. G. Wierschke, J. Chandrasekhar and W. L. Jorgensen, J. Am. Chem. Soc., 1985, 107, 1496 CrossRef CAS.
- (a) K. Itami, K. Mitsudo, T. Kamei, T. Koike, T. Nokami and J.-i. Yoshida, J. Am. Chem. Soc., 2000, 122, 12013 CrossRef CAS; (b) K. Itami, T. Nokami, Y. Ishimura, K. Mitsudo, T. Kamei and J.-i. Yoshida, J. Am. Chem. Soc., 2001, 123, 11577 CrossRef CAS PubMed; (c) K. Itami, Y. Ushiogi, T. Nokami, Y. Ohashi and J.-i. Yoshida, Org. Lett., 2004, 6, 3695 CrossRef CAS PubMed; (d) L. Ilies, J. Okabe, N. Yoshikai and E. Nakamura, Org. Lett., 2010, 12, 2838 CrossRef CAS PubMed; (e) G. Meng and M. Szostak, Angew. Chem., Int. Ed., 2015, 54, 14518 CrossRef CAS PubMed.
- No product was observed when β-bromostyrene or iodooctane was used in place of aryl bromides. Similarly, 2-pyridyldimethylallylsilane bearing olefin unactivated by Si and (E)-2-(dimethyl(3-(4-(trifluoromethyl)phenyl)allyl)silyl)pyrid-ine containing internal olefin also did not form any product.
- No product was observed when 2-propylzinc bromide, 2-pyridylzinc bromide or arylzinc iodide bearing nitrile or ester functional group was used.
- K. Itami, K. Mitsudo and J.-i. Yoshida, J. Org. Chem., 1999, 64, 8709 CrossRef CAS.
- (a) M. R. Harris, M. O. Konev and E. R. Jarvo, J. Am. Chem. Soc., 2014, 136, 7825 CrossRef CAS PubMed; (b) C. Liu, S. Tang, D. Liu, J. Yuan, L. Zheng, L. Meng and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 3638 CrossRef CAS PubMed; (c) S. A. Lebedev, V. S. Lopatina, E. S. Petrov and I. P. Beletskaya, J. Organomet. Chem., 1988, 344, 253 CrossRef CAS; (d) T. M. Gøgsig, J. Kleimark, S. O. Nilsson Lill, S. Korsager, A. T. Lindhardt, P.-O. Norrby and T. Skrydstrup, J. Am. Chem. Soc., 2012, 134, 443 CrossRef PubMed; (e) A. Trejos, J. Sävmarker, S. Schlummer, G. K. Datta, P. Nilsson and M. Larhed, Tetrahedron, 2008, 64, 8746 CrossRef CAS; (f) A. B. Machotta, B. F. Straub and M. Oestreich, J. Am. Chem. Soc., 2007, 129, 13455 CrossRef CAS PubMed; (g) J.-N. Desrosiers, L. Hie, S. Biswas, O. V. Zatolochnaya, S. Rodriguez, H. Lee, N. Grinberg, N. Haddad, N. K. Yee, N. K. Garg and C. H. Senanayake, Angew. Chem., Int. Ed., 2016, 55, 11921 CrossRef CAS PubMed; (h) K. M. M. Huihui, R. Shrestha and D. J. Weix, Org. Lett., 2017, 19, 340 CrossRef CAS PubMed.
- A. N. Abeywickrema and A. L. J. Beckwith, J. Chem. Soc., Chem. Commun., 1986, 464 RSC.
- G. D. Jones, J. L. Martin, C. McFarland, O. R. Allen, R. E. Hall, A. D. Haley, R. J. Brandon, T. Konovalova, P. J. Desrochers, P. Pulay and D. A. Vicic, J. Am. Chem. Soc., 2006, 128, 13175 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization, and the 1H and 13C NMR spectra of new compounds. See DOI: 10.1039/c7sc04351a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |