
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Qubit crossover in the endohedral fullerene Sc3C2@C80†
Zheng
Liu
a,
Bo-Wei
Dong
a,
Hai-Bing
Meng
b,
Mei-Xing
Xu
a,
Tai-Shan
Wang
 b,
Bing-Wu
Wang
a,
Chun-Ru
Wang
*b,
Shang-Da
Jiang
*a and
Song
Gao
*a
b,
Bing-Wu
Wang
a,
Chun-Ru
Wang
*b,
Shang-Da
Jiang
*a and
Song
Gao
*a
aNational Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China. E-mail: jiangsd@pku.edu.cn; gaosong@pku.edu.cn
bKey Laboratory of Molecular Nanostructure and Nanotechnology, Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: crwang@iccas.ac.cn
First published on 2nd November 2017
Abstract
The core–shell structure of endohedral fullerenes results in good protection of the encapsulated spin carriers from the environment. In this research, the quantum coherence behavior of the endohedral fullerene Sc3C2@C80 in CS2 solution is characterized from 5 K to room temperature. Below the critical temperature of around 140 K, the inner group is hindered, and the EPR spectrum consists of a single broad line. The spin carriers display a maximum phase memory time of 17.2(7) μs at 10 K. In the high temperature region, the inner group is mobile, and the EPR spectrum consists of 22 homogeneously broadened lines due to isotropic hyperfine coupling. The maximum phase memory time for each transition is around 139(1) ns at 200 K which allows arbitrary superposition state manipulations to be performed. This research demonstrates that Sc3C2@C80 displays temperature-crossover behaviour due to weak interaction between the Sc3C2 core and the C80 shell.
Introduction
Quantum computation innovates the conventional way of computing.1 The basic units for quantum computers are quantum bits (qubits). For the purpose of computation application, long phase memory times are required for quantum manipulation. Besides the various qubits involved in previous research, chemists have demonstrated that magnetic molecules can also be used as potential qubits. Several groups have successfully built qubits using paramagnetic molecules, such as Cr7Ni,2 single-molecule magnets,3,4 mononuclear transition ion complexes5–18 and endohedral fullerenes.19Endohedral fullerenes are core–shell structure molecules synthesised by encapsulating atoms in fullerene cages. This is an efficient way to isolate the core from the environment so as to stabilize the inner chemical stability and even enhance their physical properties. In previous research, the electron spin suffers rapid decoherence due to environmental effects. It is straightforward to protect the qubit in the fullerene, resulting in enhancement of the quantum coherence behavior. The carbon cage offers many advantages in comparison to other systems. The fullerene shell can work as a closed form of protection from the environment.20 Moreover, the encapsulation structure is maneuverable and visible for single molecule device manipulation.21 As an example, N@C60 is the first well-investigated endohedral fullerene qubit with a distinctive electronic structure and quantum coherence behaviour. There have been many reports on the synthesis of endohedral fullerenes.22–24
A further interesting feature of endohedral fullerenes is that the interaction between the core and shell is weak and more isotropic, indicating that motion of the inner group is possible. This is very different from traditional coordination compounds, which possess rigid chemical bonds and are normally inflexible with respect to temperature variation. It is reasonable to predict that the temperature dependence of the structure is able to induce transformation of the physical properties. A good example is spin crossover behavior which is a spin state transformation resulting from structure alternation induced by temperature and/or light.25 In previous research of molecular qubits, chemists have mainly focused on coordination compounds, whose rigid chemical bonds cause uniform quantum coherence behavior in the investigated temperature range.
Lots of carbide cluster metallofullerenes and their structures and properties have been well investigated before.26 In this study we encapsulated a Sc3C2 cluster in a C80 fullerene. Our research results demonstrate that the electron spin, in the presence of three nuclear spin carriers (45Sc, I = 7/2), is well protected in the carbon cage, resulting in a quantum phase memory time of 17.2(7) μs at 10 K which can be enhanced to 68.0(4) μs. More interestingly, this molecular qubit displays an abrupt change in quantum coherence properties upon temperature variation that arises from the core motion.
Results and Discussion
Sc3C2@C80 was originally considered as Sc3@C82,27,28 but further NMR investigation indicated that there are two types of carbon atoms in one molecule.29 The structure was first determined by single crystal X-ray diffraction analysis of the derivative molecule.29 Further investigation has provided more evidence of the structure.30 Three 45Sc ions surround the C23− group, and are encapsulated by a C80 cage (Fig. 1a). The two types of conformational isomer with the C2 lying inside and outside of the Sc3 plane have a slight energy difference.29,31 DFT calculations demonstrated that the charge distribution of the molecule can be expressed by the formula (Sc3+)3(C2)3−@(C80)6−, and the electron spin 1/2 delocalizes in the Sc3C2 cluster.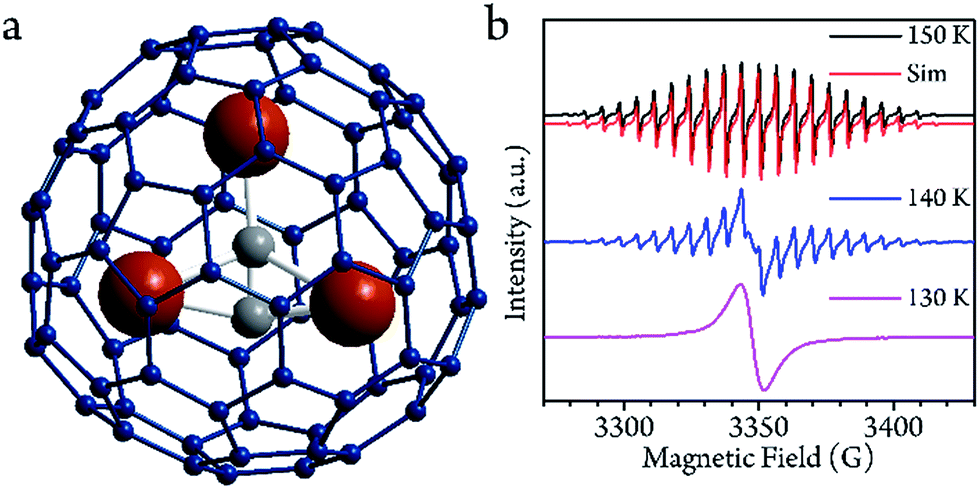 | ||
| Fig. 1 (a) The optimized structure of the Sc3C2@C80 based on DFT calculations. (b) cw-EPR spectra from 130 to 150 K in CS2 solution. The spectrum at 150 K can be reproduced with the following parameters: giso = 2.00 and Aiso = 18.10 MHz. The simulation was performed by the EasySpin toolbox in the Matlab software. | ||
cw-EPR characterization
The cw-EPR spectra of Sc3C2@C80, which were solved in a low concentration of CS2 (0.02 mmol L−1, see ESI†) from 150 K to 130 K, are plotted in Fig. 1b. To ensure the temperature stability, all of the measurements were performed after a 30 min delay once the target temperatures were achieved. At 150 K, which is below the melting point of CS2 (161 K), a 22-fold feature with a linewidth of 1.4 G could be resolved. This spectrum could be reproduced by the effective spin Hamiltonian| Ĥ = gμBBŜ + AŜÎ, |
Qubit behavior in the low temperature region
Disparate quantum coherence behaviors in the high and low temperature regions could be observed, which were also due to the core motion effect. In the low temperature region, the spin-lattice relaxation time (T1) and phase-memory time (TM) were measured by inversion recovery (π–T–π/2–τ–π–τ–echo) and Hahn echo decay, respectively. The T1 decreased upon warming across the whole temperature range, indicating that the thermal process dominates the longitudinal relaxation. On the contrary, the TM remained nearly constant below 40 K (13.6(4) μs at 5 K, a maximum of 17.2(7) μs at 10 K and 10.6(5) μs at 20 K), and started to decrease upon warming.The decoherence of the electron spin superposition state can be caused by nuclear spin, phonon or intermolecular electron dipolar interaction.36,37 According to the concentration dependence of the TM, the molarity (0.02 mmol L−1) was low enough in comparison to that of previous publications.2,7 The intermolecular dipolar–dipolar interaction is therefore regarded as a minor effect in the decoherence behavior. As to the effects of the phonon, we can consider it in two regions. At above 40 K, the TM dropped upon warming with a decrease of the T1. The TM was found to be around 1/6 of the T1. This is a strong indication that, in the temperature region above 40 K, the spin-phonon interaction was the main effect that caused decoherence of the quantum superposition. However, the Tm remained nearly constant with respect to the variation of the T1 at below 40 K. From a quantitative point of view, 1/TM is in the order of 105 s−1 which is vastly larger than that of 1/T1 (about 102 s−1). Therefore, the phonon effect contributes very little to the decoherence herein.38 The evidence leads to the conclusion that the decoherence of the electron spin at below 40 K was largely caused by surrounding nuclear spin carriers.
The nuclear spins around the electron spin act as a local magnetic field (Overhauser field, Bhf). The real magnetic field felt by the electron is the combination of the magnetic field of the spectrometer (B0) and the fluctuating Overhauser field. This effect leads to inhomogeneous broadening, and reduces the TM. Herein, we demonstrate that the fluctuating Overhauser effect is suppressed by the dynamic decoupling in the present molecule, where a train of refocusing pulses are applied before the echo detection.39 The following CPMG-n pulse sequence (π/2–τ–(π − 2τ)n−1–π–τ–echo) was applied to the system, where n denotes the number of the inversion π pulses. As previously discussed, the fluctuating Overhauser field is the major effect that causes the decoherence. Therefore, dynamic decoupling enhanced the TM effectively. When the CPMG-1 (Hahn pulses) was applied, the TM was only 13 μs. When there were more refocusing pulses, they effectively increased the TM to 67 μs with 32 π pulses. Nevertheless, we were unable to keep increasing n because the echo intensity also decreased largely at the same time. The applied pulses can never be perfect. Therefore, the error of the refocusing pulses would accumulate and finally destroy the quantum phase coherence. One solution is to employ more effective dynamic decoupling sequences.40–42
The electron spin echo envelope modulation (ESEEM) measurement was taken at 5 K (see ESI†). However, the anisotropic hyperfine interaction involving three different Sc3+ ions would result in too many parameters to determine. Moreover, the Hilbert space is of 1024-dimension, making the simulation difficult.
Qubit behavior in the high temperature region
As the system warmed up from the base temperature, the spin echo intensity decreased. At above 80 K, the intensity of the spin echo was largely reduced, was not resolvable in environmental noise and became hardly visible. Nevertheless, the spin echo signals suddenly appeared at 150 K. The echo detected field swept (EDFS) spectrum was measured at 200 K and clearly shows the 22-line feature, as shown in Fig. 2b. Every individual line in the cw-EPR spectrum is of Lorentzian shape, indicating that the TM was not affected by nuclei spin. The major effect that limited the spin coherence time was the rapid longitudinal relaxation. The TM and T1 could be recorded from 150 K to room temperature, as shown in Fig. 3b. In this temperature range, the T1 decreased as the temperature increased, while the TM showed a maximum value of 139(1) ns at around 200 K. This was probably caused by the fact that the 13C located at the outer shell with a natural abundance (1.07%) decoherence effect was averaged when the solution melted above 161 K and the fullerene cages started to rotate rapidly. This analysis reveals that the 22 transitions concerning different nuclear spin momentum behave with a similar phase memory time at a certain temperature while higher nuclear spin moments will slightly accelerate the phase decoherence. This finding is consistent with previous research.43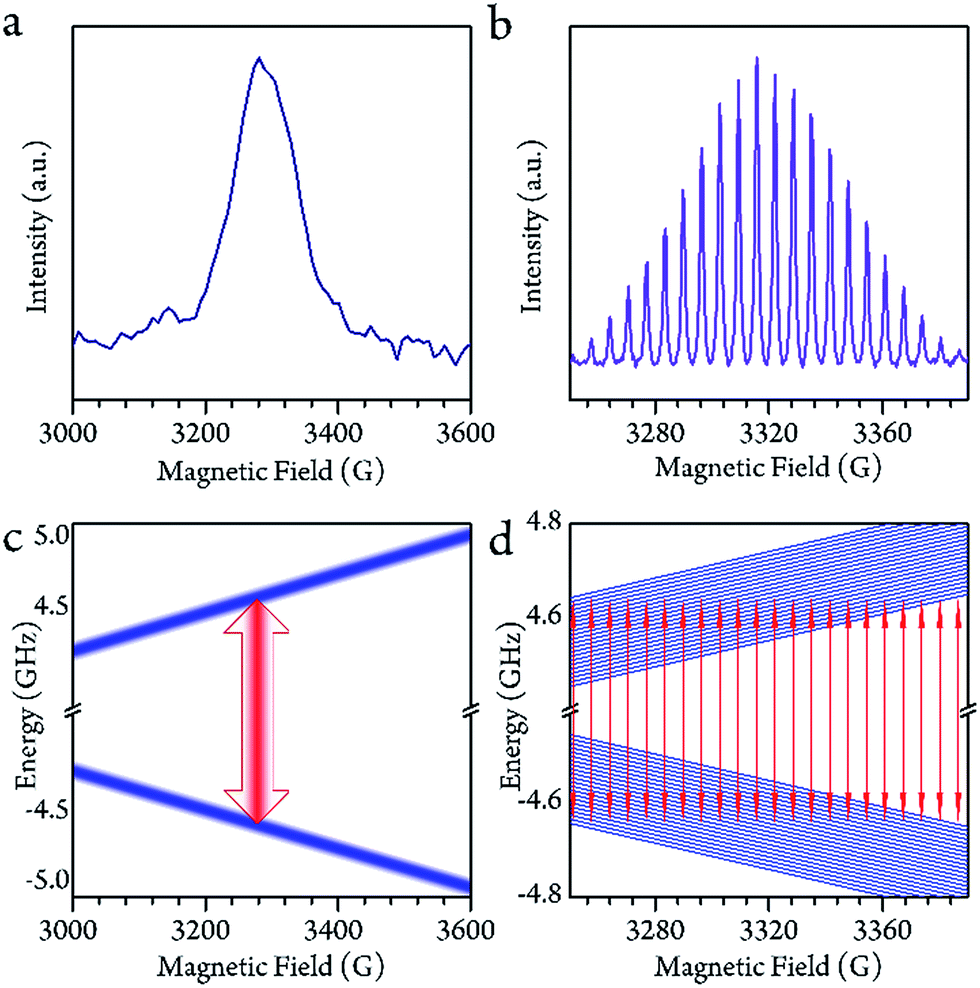 | ||
| Fig. 2 (a and b) The echo detected field swept (EDFS) spectra of the sample at 5 K and 200 K, respectively. Their corresponding energy levels and transitions are indicated in parts c and d. In the low temperature region (a and c) the energy diagram is represented as a pseudo-two-level picture; in the high temperature region (b and d), the 22-transition lines are well resolved. | ||
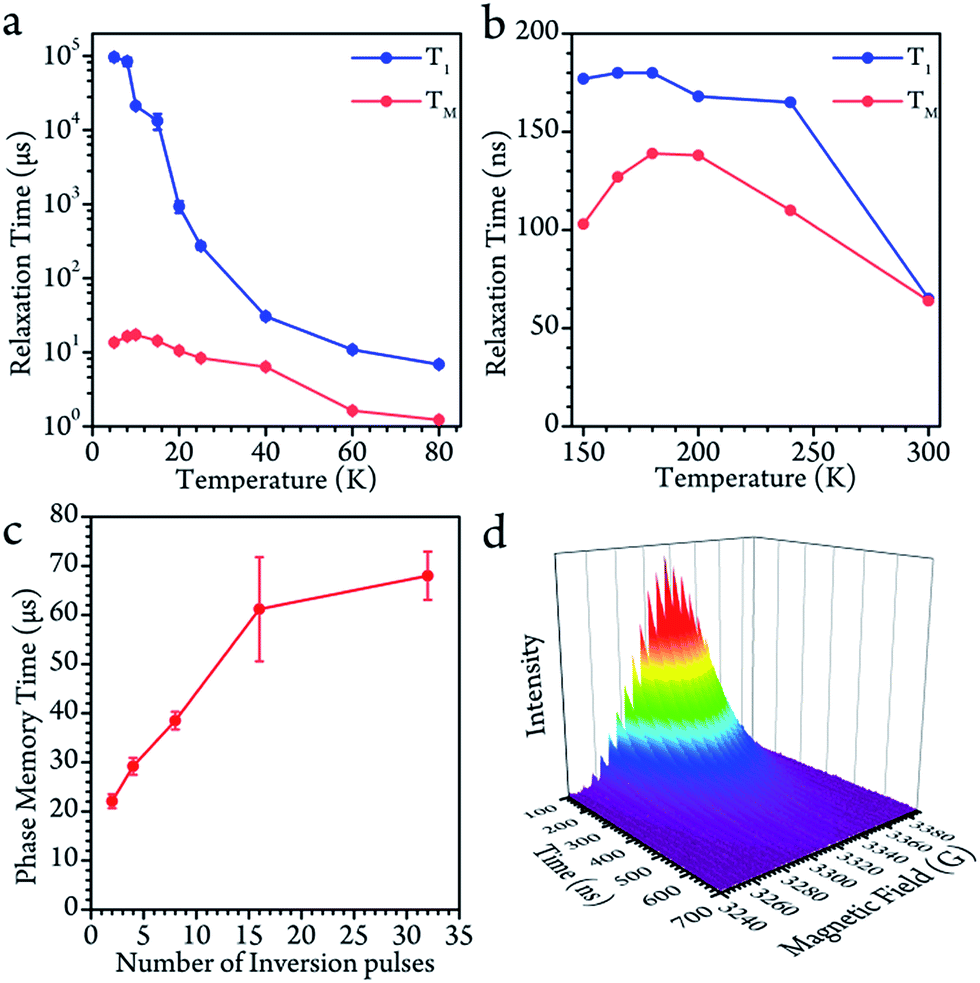 | ||
| Fig. 3 (a and b) The longitudinal relaxation time (T1) and phase memory time (TM) in the low and high temperature regions. The spin echo signal is hard to record from 80 to 150 K. (c) The phase memory time enhanced by dynamic decoupling with various numbers of inversion pulses. (d) The spin echo decays as a function of the magnetic field. | ||
Rabi oscillation
To check for the possibility of putting the spin in an arbitrary superposition state, a nutation experiment was performed in both temperature regions. In the present hyperfine coupled system, the superposition state involved only the electron spin states |±1/2〉, as the variation of the nuclear spin momentum is forbidden in the EPR transition. An oscillating magnetic field (B1) from the microwave was applied to drive the spin rotation in the plane normal to the B1 field. The tip angle (θ) depends on the applied strength and time of the B1 field.In the high temperature region, we detected magnetization of the spin, and observed that it oscillated with a certain period as the length of the driven pulse, known as Rabi oscillation. The frequency (ΩR) is proportional to the driven magnetic field.38 The nutation experiment was performed at a variety of temperatures, and the Fourier transform result is shown in Fig. 4c and the ESI.† The oscillation decays mainly resulted from decoherence of the spin. Further investigation of the field scan also showed that nutation was available for all of the transitions (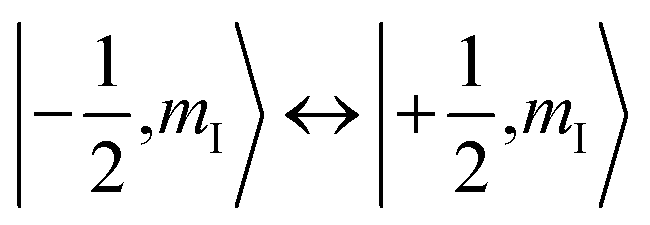 ). The whole spectrum of Sc3C2@C80 is approximately 135 G, offering the possibility to excite any transitions with an arbitrary wave generator (AWG) of 400 MHz bandwidth when the external B0 field is set to the center of the spectrum. Therefore, the arbitrary superposition state of basis
). The whole spectrum of Sc3C2@C80 is approximately 135 G, offering the possibility to excite any transitions with an arbitrary wave generator (AWG) of 400 MHz bandwidth when the external B0 field is set to the center of the spectrum. Therefore, the arbitrary superposition state of basis 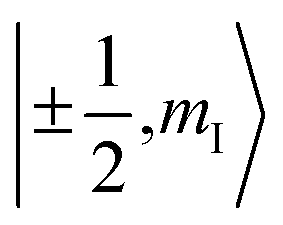 with any specific mI value can be manipulated without varying the B0 field.
with any specific mI value can be manipulated without varying the B0 field.
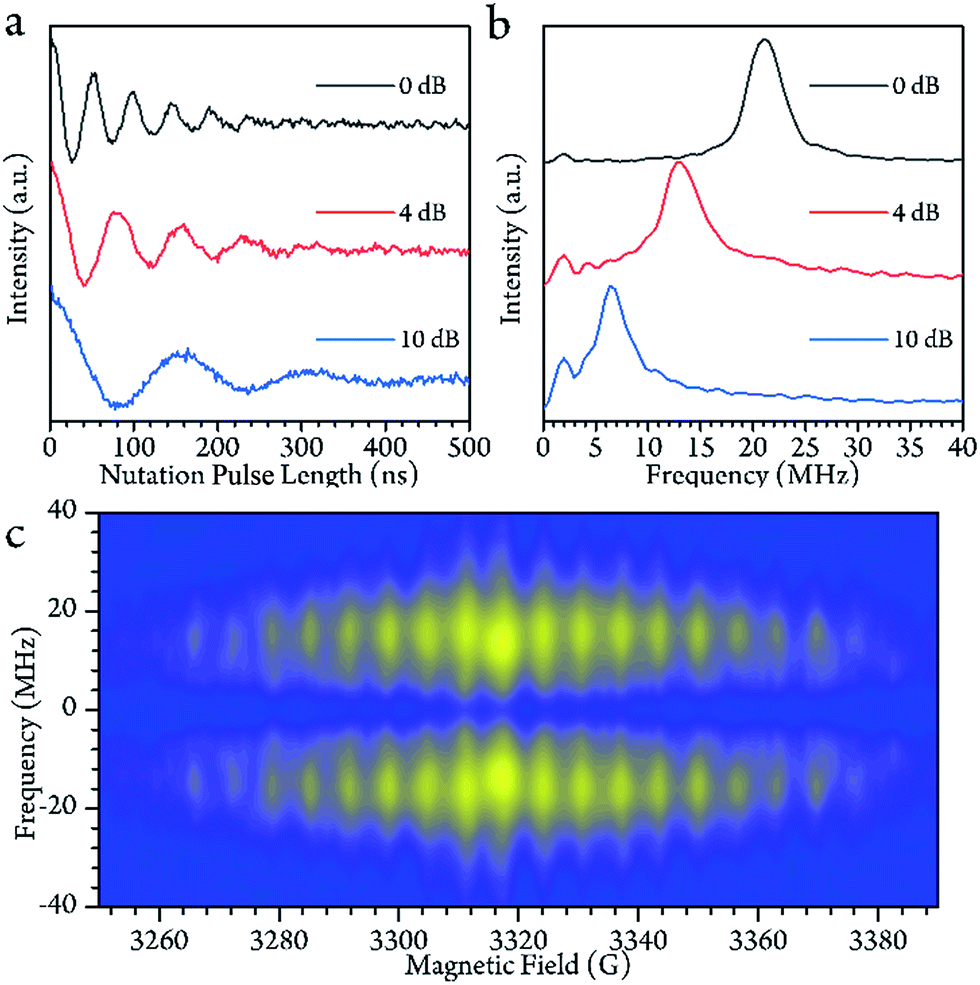 | ||
| Fig. 4 (a) The Rabi oscillation measured at the center of the field at 200 K with various powers of microwaves. The value of dB indicates the microwave attenuated from the full power (300 W) of the solid-state amplifier. (b) The Fourier transform of (a). The oscillation frequency behaves as a linear function of the microwave B1 field. (c) The Fourier-transformed Rabi oscillation as a function of the magnetic field B0 at 200 K. The Rabi frequencies remain constant over the whole field range. | ||
In the low temperature region, however, the Rabi oscillation is hardly visible. This is probably due to the degrees of freedom with respect to the electron and nuclear being strongly linked to each other since the core motion is frozen at below 140 K. This situation changes when the core mobility is obvious.
Conclusion
Different from previously reported molecular qubits, Sc3C2@C80 is a dual-behavioral qubit. Its quantum coherence behaviors are very different below and above the critical temperature of around 140 K. In the low temperature region, the longest quantum phase memory time is nearly 20 μs. However, only one broad line is resolved and Rabi oscillation is hardly observable. In contrast, all of the 22 transitions’ quantum coherence is evidently observed and the arbitrary superposition state of the |±1/2〉 basis with any specific nuclear spin momentum is accessible in the high temperature region. The phase memory time at high temperature is two orders shorter than that of the low temperature region. Considering the weak polarization, this crossover cannot be explained by the thermal population variation of the energy levels, but is probably due to the mobility of the inner group. Since the rotational correlation time of the magnetic core Sc3C2@C80 (3.5 ps)35 is very short and the molecule possesses a large mass compared to that of dinuclear gas molecules, the spin-rotational interaction induced EPR line splitting is not observable.44 However, this environmental effect originating from the core rotation indeed causes decoherence of the quantum phase and dramatically increases upon warming.For the purpose of quantum information processing, the quantum gates are of fundamental importance, in which the controlled-NOT gate requires coupled qubits, either between electron–electron or electron–nuclear spin. However, this type of coupling could lead to rapid decoherence. By encapsulating the electron and nuclear spin carriers in the fullerene, the qubit is very well protected by the carbon cage. In the presence of three surrounding large nuclear spins, the quantum coherence can still survive for tens of microseconds, proving that the strategy is effective. Besides, the 22 transitions can be well resolved in the high temperature region. The short phase memory time is mainly limited by longitudinal relaxation. Strategies to enhance the T1 are then demanded.
Taking advantage of the carbon cage, endohedral fullerenes can be placed on the surface of or inside the carbon nanotube to assemble in dimensions. It is therefore reasonable to believe that Sc3C2@C80 could act as elements of quantum information processing, building up to quantum gates by employing the electron spin and the relevant scandium nuclei spins. For further investigation, pulse ENDOR can be used to generate an entangled state and quantum gate. An individual molecule investigation on the surface to build up quantum devices is also of general interest.
Methods
Synthesis
Sc3C2@C80 was synthesized and purified as in literature methods.27EPR measurements
The Sc3C2@C80 was dissolved in a CS2 solution for EPR experiments with a concentration of 0.02 mmol L−1 determined by the Bruker spin counting method (see ESI†). cw-EPR spectra were measured on a Bruker Elexsys E580 spectrometer with a super-high sensitivity probe head (ω = 9.37 GHz). Pulsed EPR data were collected on the same system by an MS-3 cavity (ω = 9.28 GHz). The cw-EPR spectra were simulated by the EasySpin toolbox based on Matlab. The low-temperature environment was achieved by liquid helium cryostats (ESR900 for cw and CF935 for pulse) produced by Oxford Instruments. The measurement temperatures were stabilized for 30 min before the measurements were carried out. The signal of the pulsed-EPR experiments was collected by integrating the Hahn echo (π/2–τ–π–τ–echo) with τ = 100 and 150 ns at a high and low temperature range, respectively. The T1 values were measured by the inversion recovery method (π–T–π/2–τ–π–τ–echo) with 16-step phase cycling. The Tm values were obtained by increasing the τ value of the Hahn echo sequence with 16-step phase cycling. Dynamic decoupling measurements were carried out by the CPMG sequence (πx/2–τ–(πy − 2τ)n–πy–τ–echo) with 16-step phase cycling. The π/2 and π pulse lengths in EDFS, T1 and Tm measurements, were 8 and 16 ns, respectively. Nutation experiments were carried out with a standard sequence (tp–T–π/2–τ–π–τ–echo), where T > 5Tm. The π/2 pulse lengths were adjusted to 10, 16 and 32 ns by 0 dB, 4 dB and 10 dB attenuation.Author contributions
Z. L. and B.-W. D. performed the EPR measurements, assisted by M.-X. X. Z. L. analyzed the EPR data. H.-B. M. synthesized the sample in support of T.-S. W. and C.-R. W. The project was conceived by S. G. and S.-D. J. S.-D. J. designed the experiments and wrote the manuscript. All of the authors revised the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research is supported by the National Natural Science Foundation of China (grant no. 21621061 and 21601005), the National Basic Research Program of China (grant no. 2017YFA0204903), and the Young Elite Scientist Sponsorship Program of the China Association of Science and Technology (grant no. YESS20150011).References
- R. P. Feynman, Int. J. Theor. Phys., 1982, 21, 467–488 CrossRef.
- A. Ardavan, O. Rival, J. J. L. Morton, S. J. Blundell, A. M. Tyryshkin, G. A. Timco and R. E. P. Winpenny, Phys. Rev. Lett., 2007, 98, 057201 CrossRef PubMed.
- C. Schlegel, J. van Slageren, M. Manoli, E. K. Brechin and M. Dressel, Phys. Rev. Lett., 2008, 101, 147203 CrossRef CAS PubMed.
- S. Takahashi, I. S. Tupitsyn, J. van Tol, C. C. Beedle, D. N. Hendrickson and P. C. E. Stamp, Nature, 2011, 476, 76–79 CrossRef CAS PubMed.
- M. S. Fataftah, J. M. Zadrozny, S. C. Coste, M. J. Graham, D. M. Rogers and D. E. Freedman, J. Am. Chem. Soc., 2016, 138, 1344–1348 CrossRef CAS PubMed.
- M. J. Graham, J. M. Zadrozny, M. Shiddiq, J. S. Anderson, M. S. Fataftah, S. Hill and D. E. Freedman, J. Am. Chem. Soc., 2014, 136, 7623–7626 CrossRef CAS PubMed.
- J. M. Zadrozny, J. Niklas, O. G. Poluektov and D. E. Freedman, ACS Cent. Sci., 2015, 1, 488–492 CrossRef CAS PubMed.
- J. M. Zadrozny, J. Niklas, O. G. Poluektov and D. E. Freedman, J. Am. Chem. Soc., 2014, 136, 15841–15844 CrossRef CAS PubMed.
- C. J. Yu, M. J. Graham, J. M. Zadrozny, J. Niklas, M. D. Krzyaniak, M. R. Wasielewski, O. G. Poluektov and D. E. Freedman, J. Am. Chem. Soc., 2016, 138, 14678–14685 CrossRef CAS PubMed.
- M. J. Graham, C. J. Yu, M. D. Krzyaniak, M. R. Wasielewski and D. E. Freedman, J. Am. Chem. Soc., 2017, 139, 3196–3201 CrossRef CAS PubMed.
- M. Atzori, L. Tesi, E. Morra, M. Chiesa, L. Sorace and R. Sessoli, J. Am. Chem. Soc., 2016, 138, 2154–2157 CrossRef CAS PubMed.
- M. Atzori, E. Morra, L. Tesi, A. Albino, M. Chiesa, L. Sorace and R. Sessoli, J. Am. Chem. Soc., 2016, 138, 11234–11244 CrossRef CAS PubMed.
- L. Tesi, E. Lucaccini, I. Cimatti, M. Perfetti, M. Mannini, M. Atzori, E. Morra, M. Chiesa, A. Caneschi, L. Sorace and R. Sessoli, Chem. Sci., 2016, 7, 2074–2083 RSC.
- M. Atzori, L. Tesi, S. Benci, A. Lunghi, R. Righini, A. Taschin, R. Torre, L. Sorace and R. Sessoli, J. Am. Chem. Soc., 2017, 139, 4338–4341 CrossRef CAS PubMed.
- K. Bader, D. Dengler, S. Lenz, B. Endeward, S. D. Jiang, P. Neugebauer and J. van Slageren, Nat. Commun., 2014, 5, 6304 Search PubMed.
- K. Bader, M. Winkler and J. van Slageren, Chem. Commun., 2016, 52, 3623–3626 RSC.
- M. J. Martinez-Perez, S. Cardona-Serra, C. Schlegel, F. Moro, P. J. Alonso, H. Prima-Garcia, J. M. Clemente-Juan, M. Evangelisti, A. Gaita-Arino, J. Sese, J. van Slageren, E. Coronado and F. Luis, Phys. Rev. Lett., 2012, 108, 247213 CrossRef CAS PubMed.
- K. S. Pedersen, A. M. Ariciu, S. McAdams, H. Weihe, J. Bendix, F. Tuna and S. Piligkos, J. Am. Chem. Soc., 2016, 138, 5801–5804 CrossRef CAS PubMed.
- S. C. Benjamin, A. Ardavan, G. A. D. Briggs, D. A. Britz, D. Gunlycke, J. Jefferson, M. A. G. Jones, D. F. Leigh, B. W. Lovett, A. N. Khlobystov, S. A. Lyon, J. J. L. Morton, K. Porfyrakis, M. R. Sambrook and A. M. Tyryshkin, J. Phys.: Condens. Matter, 2006, 18, S867–S883 CrossRef CAS.
- T. A. Murphy, T. Pawlik, A. Weidinger, M. Hohne, R. Alcala and J. M. Spaeth, Phys. Rev. Lett., 1996, 77, 1075–1078 CrossRef CAS PubMed.
- A. N. Khlobystov, K. Porfyrakis, M. Kanai, D. A. Britz, A. Ardavan, H. Shinohara, T. J. S. Dennis and G. A. D. Briggs, Angew. Chem., Int. Ed., 2004, 43, 1386–1389 CrossRef CAS PubMed.
- D. S. Bethune, R. D. Johnson, J. R. Salem, M. S. de Vries and C. S. Yannoni, Nature, 1993, 366, 123–128 CrossRef CAS.
- A. Rodriguez-Fortea, A. L. Balch and J. M. Poblet, Chem. Soc. Rev., 2011, 40, 3551–3563 RSC.
- X. Lu, L. Feng, T. Akasaka and S. Nagase, Chem. Soc. Rev., 2012, 41, 7723–7760 RSC.
- A. Bousseksou, G. Molnar, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313–3335 RSC.
- X. Lu, T. Akasaka and S. Nagase, Acc. Chem. Res., 2013, 46, 1627–1635 CrossRef CAS PubMed.
- H. Shinohara, H. Sato, M. Ohkohchi, Y. Ando, T. Kodama, T. Shida, T. Kato and Y. Saito, Nature, 1992, 357, 52–54 CrossRef CAS.
- A. Bartl and L. Dunsch, Synth. Met., 2001, 121, 1147–1148 CrossRef CAS.
- Y. Iiduka, T. Wakahara, T. Nakahodo, T. Tsuchiya, A. Sakuraba, Y. Maeda, T. Akasaka, K. Yoza, E. Horn, T. Kato, M. T. Liu, N. Mizorogi, K. Kobayashi and S. Nagase, J. Am. Chem. Soc., 2005, 127, 12500–12501 CrossRef CAS PubMed.
- H. Fang, H. Cong, M. Suzuki, L. Bao, B. Yu, Y. Xie, N. Mizorogi, M. M. Olmstead, A. L. Balch, S. Nagase, T. Akasaka and X. Lu, J. Am. Chem. Soc., 2014, 136, 10534–10540 CrossRef CAS PubMed.
- K. Tan and X. Lu, J. Phys. Chem. A, 2006, 110, 1171–1176 CrossRef CAS PubMed.
- J. Rahmer, L. Dunsch, H. Dorn, J. Mende and M. Mehring, Magn. Reson. Chem., 2005, 43, S192–S198 CrossRef CAS PubMed.
- H. Shinohara, M. Inakuma, N. Hayashi, H. Sato, Y. Saito, T. Kato and S. Bandow, J. Phys. Chem., 1994, 98, 8597–8599 CrossRef CAS.
- K. Tan and X. Lu, J. Phys. Chem. A, 2006, 110, 1171–1176 CrossRef CAS PubMed.
- S. Taubert, M. Straka, T. O. Pennanen, D. Sundholm and J. Vaara, Phys. Chem. Chem. Phys., 2008, 10, 7158–7168 RSC.
- W. Wernsdorfer, Nat. Mater., 2007, 6, 174–176 CrossRef CAS PubMed.
- A. Morello, P. C. E. Stamp and I. S. Tupitsyn, Phys. Rev. Lett., 2006, 97, 207206 CrossRef PubMed.
- A. Schweiger and G. Jeschke, Principles of Pulse Electron Paramagnetic Resonance, Oxford University Press, 2001 Search PubMed.
- S. Meiboom and D. Gill, Rev. Sci. Instrum., 1958, 29, 688–691 CrossRef CAS.
- G. S. Uhrig, Phys. Rev. Lett., 2007, 98, 100504 CrossRef PubMed.
- M. J. Biercuk, H. Uys, A. P. VanDevender, N. Shiga, W. M. Itano and J. J. Bollinger, Nature, 2009, 458, 996–1000 CrossRef CAS PubMed.
- J. F. Du, X. Rong, N. Zhao, Y. Wang, J. H. Yang and R. B. Liu, Nature, 2009, 461, 1265–1268 CrossRef CAS PubMed.
- J. J. L. Morton, A. M. Tyryshkin, A. Ardavan, K. Porfyrakis, S. A. Lyon and G. A. D. Briggs, J. Chem. Phys., 2006, 124, 014508 CrossRef PubMed.
- I. B. Goldberg and H. O. Laeger, J. Phys. Chem., 1980, 84, 3040–3045 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc03749j |
| This journal is © The Royal Society of Chemistry 2018 |