
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCalcium-mediated one-pot preparation of isoxazoles with deuterium incorporation†
Maria S.
Ledovskaya
a,
Konstantin S.
Rodygin
a and
Valentine P.
Ananikov
 *ab
*ab
aSaint Petersburg State University, Universitetsky prospect 26, Peterhof, 198504, Russia
bN. D. Zelinsky Institute of Organic Chemistry Russian Academy of Sciences, Leninsky prospect 47, Moscow, 119991, Russia. E-mail: val@ioc.ac.ru
First published on 28th September 2017
Abstract
In this work, a novel synthetic methodology for the one-pot preparation of isoxazoles directly from the reaction of calcium carbide with aldoximes is reported. Calcium carbide acts as a safe and inexpensive acetylene source and, in addition, as a source of the Ca(OH)2 base to enable the generation of nitrile oxide. Various 3-substituted isoxazoles were synthesized from the corresponding aldoximes in good yields (up to 95%) and a series of new deuterated 4,5-dideuteroisoxazoles were prepared.
Introduction
The covalent carbon–deuterium (C–D) bond is noticeably stronger compared to a carbon–hydrogen (C–H) bond. This difference in the bond strength opens many practical opportunities. For example, comparison of the C–H bond and C–D bond reactivity may be used in the study of reaction mechanisms.1 In addition to mechanistic studies, deuterated starting materials can be applied to alter the reaction selectivities in total syntheses. This approach has been utilized in the synthesis of pharmaceutical substances.2 Recently, deuterium substitution led to a very interesting application with the enhancement of the metabolic stability of drugs. The larger strength of the C–D bond in deuterated pharmaceutical substances leads to a higher effectiveness and lower toxicity in comparison with those of the undeuterated analogs.3 In view of the recent advances in this field,4 the synthesis of new deuterated substances is a demanded goal for organic and pharmaceutical chemistry.Heterocyclic molecules possess a diverse range of biological activities and are ubiquitously used in many drugs.3–5 Undoubtedly, the deuteration of heterocyclic cores is a demanded research area to develop a new generation of drugs. Isoxazoles demonstrate a wide range of biological activities and are used in drug development.6 However, all the methods used previously to obtain deuterated isoxazoles had severe limitations. In particular, deuterium sulfate or deuterium chloride was used to introduce deuterium into the isoxazole ring. Using this procedure, the starting isoxazole was treated under harsh conditions with acidic reagents to obtain 4- or 5-deuteroisoxazoles (Scheme 1, path a).7 This approach has rather limited functional group tolerance.
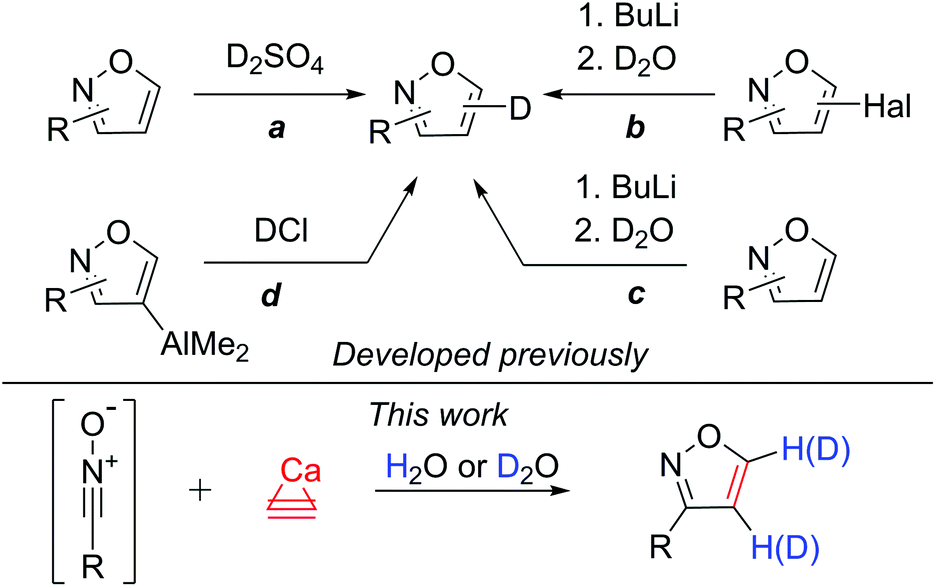 | ||
| Scheme 1 Known deuteration via the substitution reaction and the developed atom-economical route via cycloaddition. | ||
The other way to yield 4- or 5-deuteroisoxazoles is the sequential treatment of 4- and 5-halogenated isoxazoles with butyl lithium and deuterium oxide (Scheme 1, path b).8 Additionally, 4- or 5-deuteroisoxazoles can be obtained by lithium-mediated H-D exchange (Scheme 1, path c).1a,b The application of BuLi as a reagent imposes even more difficulties with regard to the functional group tolerance. Highly air- and moisture-sensitive aluminated isoxazoles can be used to prepare 4-deuteroisoxazoles by reaction with DCl under glovebox conditions (Scheme 1, path d).9 A few other approaches to deuteroisoxazoles have also been reported.10
In this work, we propose a novel synthetic approach to prepare isoxazoles with the facile ability for deuterium incorporation. This methodology is based on a 1,3-dipolar cycloaddition of nitrile oxide to acetylene or deuteroacetylene generated in situ (Scheme 1). Here, calcium carbide acts as a safe and readily available acetylene source.11 Nitrile oxide is generated from the corresponding chloroaldoximes in the presence of a base.12 Chloroaldoximes can be easily synthesized by aldoxime chlorination with chlorine,13N-chlorosuccinimide (NCS)14 or other reagents.15
Results and discussion
As a model process, the reaction of 4-tolualdehyde oxime 1a and chlorinated derivative 2a with calcium carbide was chosen. To test the proposed utilization of calcium carbide as an acetylene source, we first studied the sequential transformation (Table 1). Compound 2a was synthesized from 1a and isolated, followed by a series of experiments utilizing 2a as a starting material. First, we tested various solvents (entries 1–6, Table 1) and obtained the best result with carbon tetrachloride (entry 6, Table 1). Using triethylamine and pyridine as bases gave the same yields (entries 6 and 7, Table 1).|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Solvent | Base | Yield, % |
| a Reaction conditions: 1a or 2a (0.3 mmol), CaC2 (2.0 mmol), H2O (4.0 mmol), N-chlorosuccinimide (0.4 mmol, for reactions with 1a), base (0.3 mmol), solvent (1.5 ml), 20 °C, 48 h; yields determined by NMR. b 3.0 mmol of CaC2 and 6.0 mmol of H2O were used. c 1.0 mmol of CaC2 and 2.0 mmol of H2O were used. | ||||
| 1 | 2a | DMSO | Et3N | 28 |
| 2 | 2a | Et2O | Et3N | 40 |
| 3 | 2a | Benzene | Et3N | 81 |
| 4 | 2a | CH2Cl2 | Et3N | 77 |
| 5 | 2a | CHCl3 | Et3N | 75 |
| 6 | 2a | CCl4 | Et3N | 82 |
| 7 | 2a | CCl4 | Py | 81 |
| 8 | 2a | CCl4 | — | 82 |
| 9b | 2a | CCl4 | — | 80 |
| 10c | 2a | CCl4 | — | 66 |
| 11 | 1a | CCl4 | — | 82 |
| 12 | 1a | CCl4 | Et3N | 82 |
| 13 | 1a | CCl4 | Py | 80 |
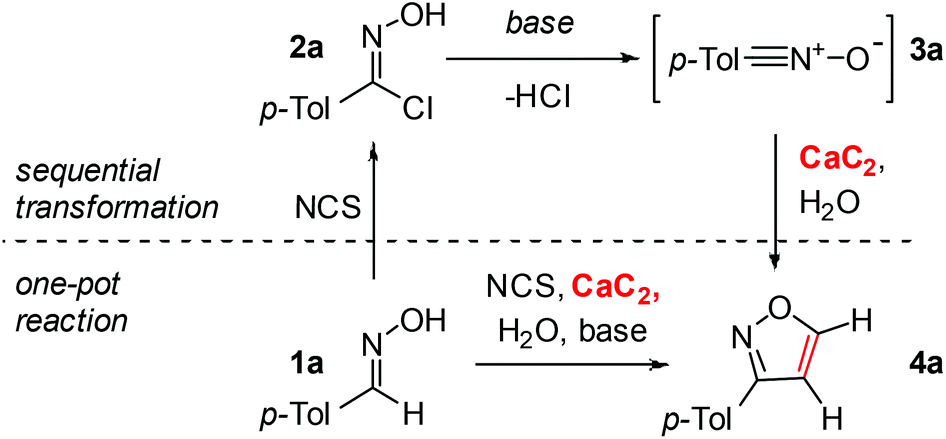
Surprisingly, Ca(OH)2 formed in situ upon reaction of CaC2 with water was an excellent base in this process, and the resulting isoxazole was formed in 82% yield without the addition of any extra base (entry 8, Table 1). The variation in the amount of calcium carbide revealed that 2 mmol was a suitable value (entries 8–10, Table 1). It should be noted that the reaction proceeded in a good yield using 1 mmol of calcium carbide (entry 10, Table 1), however an excess of this reagent improved the selectivity by suppressing the side-reactions. Taking into account the very low price of calcium carbide and the nontoxic nature of generated calcium hydroxide, the usage of an excess of calcium carbide is the synthetic procedure of choice for carrying out the reaction.
Next, we addressed the possibility of a one-pot reaction to synthesize 4a directly from 1a (entry 11, Table 1). For this transformation N-chlorosuccinimide was used to generate 2ain situ. The yield in the one-pot reaction of 1a with NCS and calcium carbide was identical compared to that of the corresponding sequential process (entries 11 and 8, Table 1). The application of Ca(OH)2 as a base was confirmed for the one-pot reaction as well, where the addition of an extra base did not change the product yield (entries 11 and 12, Table 1).
With the optimized conditions in hand, the substrate scope with a variety of aldoximes was explored (Table 2). The reaction worked well for a wide range of substrates with various functional groups, affording the desired isoxazoles in moderate to excellent yields. The highest yields were observed for the reactions of calcium carbide with 1b (95% yield), 1c and 1e (91% and 92% yields), ortho-iodo- (1g) and ortho-bromo- (1h) derivatives (95% and 93% yields), 1t and 1u (93% and 94% yields). Benzaldoxime 1f gave 4f in good yield (84%), and aldoximes 1a, 1i–1k, 1m, 1o, and 1r also provided good yields (72–87%). Anthracene derivative 1x and substrate 1y derived from (1R)-(−)-myrtenal gave the desired isoxazoles in good yields (88% and 82%, respectively). Overall, a facile transformation was developed for the preparation of many products (Table 2).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: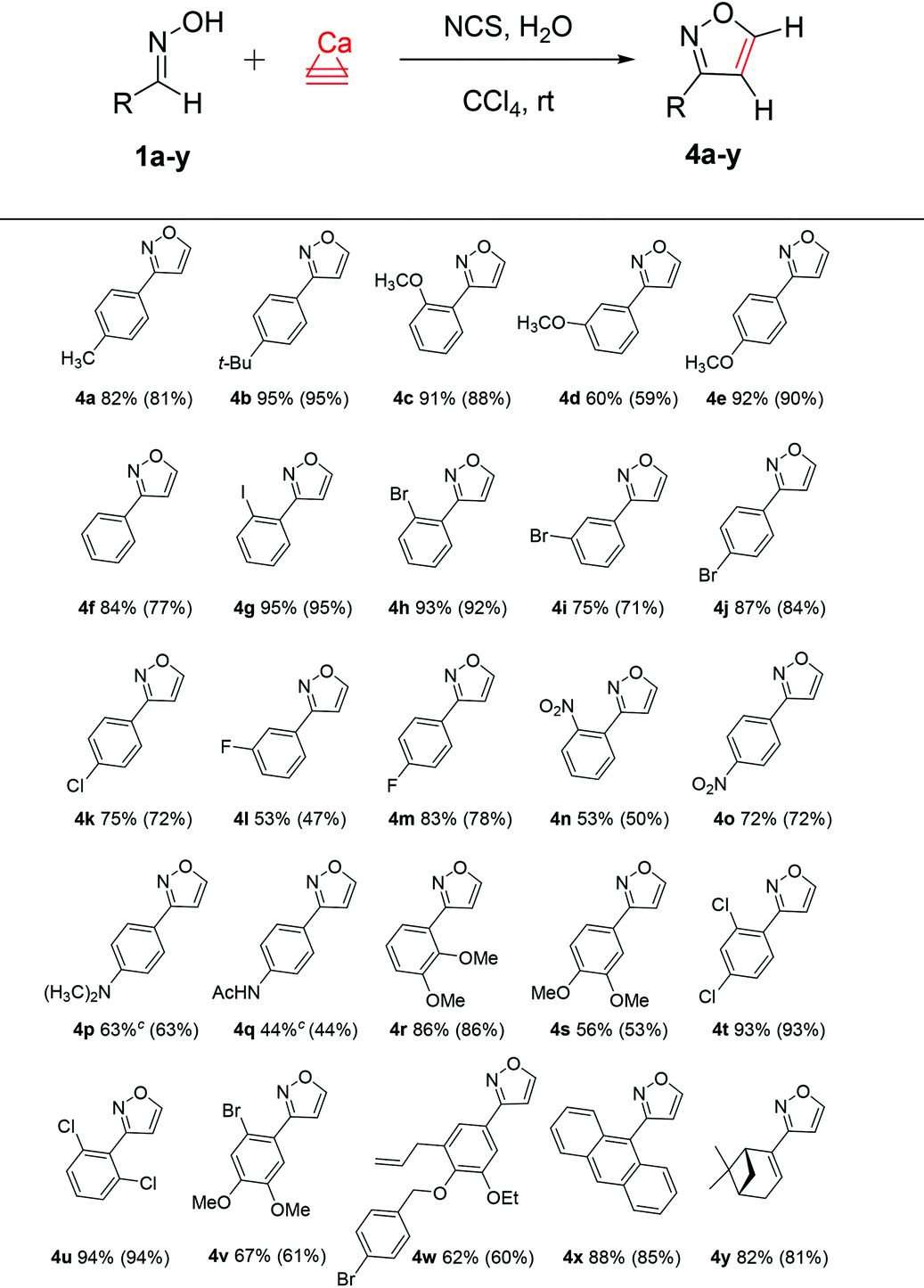
Moderate yields of nitro-, dimethylamino- and acetamido-derived products 4n–4q were related to the poor solubility of the starting materials in the organic solvent. For the synthesis of 4p and 4q, we tested other solvents (CH2Cl2, CHCl3, CCl4, benzene, C6H6–CH2Cl2, and CCl4–EtOAc), and a C6H6–CH2Cl2 mixture provided better isolated product yields of 63% and 44%, respectively.
For additional characterization, product 4z was synthesized and the structure was established by X-ray analysis (see the ESI†).
The application of calcium carbide provides an excellent possibility for deuterium incorporation. Deuterated acetylene C2D2 was generated by the reaction of CaC2 with D2O. The optimization of the deuteration procedure involving varying the amounts of D2O and the reagents, as well as changing the reaction conditions, was carried out (entries 1–8, Table 3).16 As a result, an excellent overall deuterium incorporation of 98% was observed under the optimized conditions (entry 9, Table 3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | D2O (mmol) | Product distribution | Deuterium incorporationc, % | ||
| 5 , % | 5′ + 5′′b, % | 4 , % | |||
| a Reaction conditions: Aldoxime 1u (R = 2,6-Cl2C6H3, 0.3 mmol), CaC2 (2.0 mmol), D2O (4.0 mmol), N-chlorosuccinimide (0.4 mmol), CCl4 (1.5 ml), 20 °C, 48 h. b Determined by NMR according to the intensity of the residual 1H signals. c Overall deuterium incorporation into products = 5 + (5′ + 5′′)/2. d The two-step sequence with succinimide removal. e The starting oxime was treated with D2O before the reaction with CaC2. f The starting oxime was treated with a D2O–CDCl3 mixture. g 1.5 mmol of CaC2 was used. h Starting aldoxime (0.25 mmol) and NCS (0.3 mmol), with all other parameters the same as in entry 8. | |||||
| 1 | 2 | 80 | 18 | 2 | 89 |
| 2 | 4 | 82 | 16 | 2 | 90 |
| 3 | 8 | 86 | 12 | 2 | 92 |
| 4 | 10 | 82 | 16 | 2 | 90 |
| 5 | 8d | 75 | 22 | 3 | 86 |
| 6 | 8e | 86 | 12 | 2 | 92 |
| 7 | 8f | 89 | 10 | 1 | 94 |
| 8 | 8f,g | 92 | 7 | 1 | 96 |
| 9 | 8h | 96 | 4 | Trace | 98 |
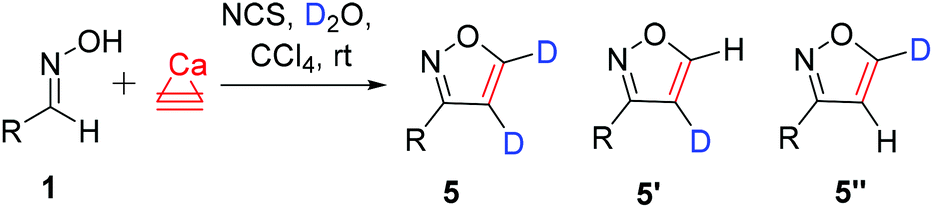
Using these optimized conditions, the deuteration process was studied for different products (Table 4). In most cases, ≥97% deuterium incorporation was observed. In only a few cases, 94–96% overall deuterium incorporation in the 4- and 5-positions of the isoxazoles was found, which nevertheless are also high-performance deuteration processes (Table 4). Synthesized 4,5-dideuteroisoxazoles 5 were stable compounds and were easily isolated by standard procedures. Purification on silica was performed without a change in the deuteration purity (see the ESI†).
| Entry | R | Yield of 5, % | Deuterium incorporationa, % |
|---|---|---|---|
| 1 | 4-MeC6H4 | 82 | 98 |
| 2 | 2-MeOC6H4 | 91 | 98 |
| 3 | 3-MeOC6H4 | 60 | 96 |
| 4 | 4-MeOC6H4 | 92 | 96 |
| 5 | Ph | 84 | 94 |
| 6 | 2-IC6H4 | 95 | 98 |
| 7 | 2-BrC6H4 | 93 | 98 |
| 8 | 3-BrC6H4 | 75 | 96 |
| 9 | 4-BrC6H4 | 87 | 98 |
| 10 | 4-ClC6H4 | 75 | 98 |
| 11 | 3-FC6H4 | 53 | 96 |
| 12 | 4-FC6H4 | 83 | 98 |
| 13 | 2,3-(MeO)2C6H3 | 86 | 97 |
| 14 | 3,4-(MeO)2C6H3 | 56 | 98 |
| 15 | 2,4-Cl2C6H3 | 93 | 97 |
| 16 | 2,6-Cl2C6H3 | 94 | 98 |
| 17 | Anthracene-9-yl | 88 | 97 |
| 18 |

|
82 | 97 |
It was of much interest to investigate the reaction mechanism. First, the key role of calcium hydroxide formed in situ was studied (Scheme 2). Two reactions were carried out using a solution saturated with acetylene gas (an external source of acetylene gas was used without the addition of CaC2). To one reaction, the corresponding amount of Ca(OH)2 was added, and both reactions were conducted under the same conditions at 20 °C for 48 h. Indeed, product formation was observed only in the presence of Ca(OH)2.
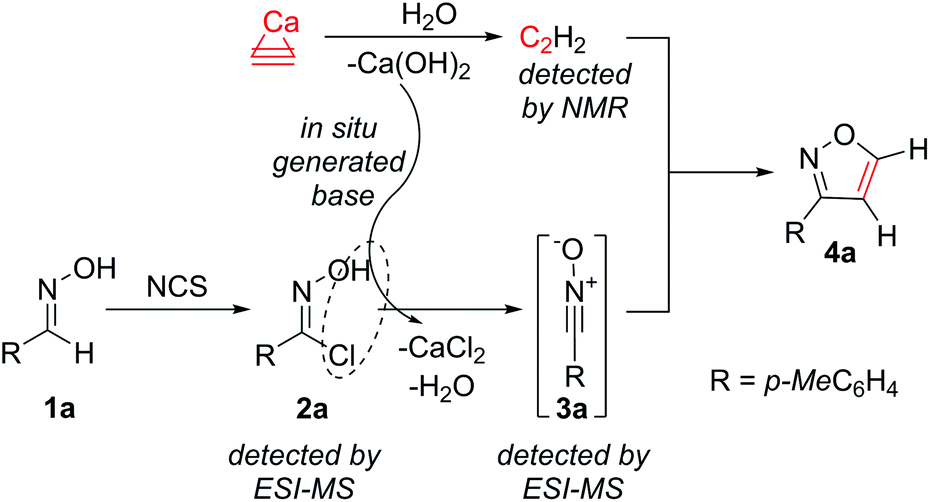 | ||
| Scheme 2 Proposed mechanism of isoxazole formation. | ||
The generation of acetylene in the studied system was confirmed by NMR (Scheme 2). Reaction monitoring with electrospray ionization mass-spectrometry (ESI-MS) helps in detecting the corresponding peaks for chloroaldoxime 2a (m/z = 170.0369 [M + H]+) and nitrile oxide 3a (m/z = 156.0416, [M + Na]+). The advantage of the developed procedure was the controlled reaction of calcium carbide with water, thus gradually releasing gaseous acetylene and calcium hydroxide. A small concentration of reactive intermediate 3a provided reliable conditions for the reaction of interest and avoided side reactions.
Conclusions
To summarize, a simple procedure for the preparation of 3-substituted isoxazoles and novel 4,5-dideuteroisoxazoles was developed. This procedure was based on the reaction of readily available aldoximes and N-chlorosuccinimide with calcium carbide. Calcium carbide acted as a safe and inexpensive source of acetylene (C2H2 and C2D2) and provided Ca(OH)2 to mediate the reaction. A nontoxic inorganic salt, CaCl2, was released at the end of the reaction, and the products were easily separated. The replacement of acetylene with CaC2 allowed for the use of standard laboratory equipment (without complicated equipment for high-pressure acetylene) and for the easy incorporation of deuterium.A combination of CaC2 and D2O is an attractive option for deuterium incorporation via atom-economical cycloaddition reactions involving C2D2. Deuterium labeling via cycloaddition has valuable advantages compared to substitution reactions, which are not atom economical and possess poor functional group tolerance. The studied route for in situ C2D2 generation has a valuable advantage in terms of not only a convenient experimental procedure, but also cost-efficiency (C2D2 gas in balloons is highly expensive). We anticipate that the application of this protocol will provide access to other valuable heterocyclic and labelled molecules.
Experimental
General procedure for the synthesis of aldoximes 1
To a round-bottom flask equipped with a magnetic stir bar, 0.02 mol of the corresponding aldehyde and 0.022 mol of hydroxylamine hydrochloride were added. Then, 15 ml of ethanol and 5 ml of water were added, followed by 0.015 mol of sodium carbonate (added in one portion). The resulting mixture was stirred at room temperature for 24 hours. Then, the solvent was evaporated, and the residue was washed with water to remove inorganics and recrystallized from the appropriate solvent.Sequential reactions for the synthesis of 3-substituted isoxazoles 4 from chloroaldoximes and calcium carbide
The corresponding chloroaldoximes were synthesized by the procedure reported previously.17 A reaction tube was loaded with chloroaldoxime (0.3 mmol) and calcium carbide (2 mmol). Then, 1.5 ml of CCl4 (or toluene) was added, the resulting mixture was stirred until a suspension formed, and deionized water (4 mmol) was added. The reaction tube was immediately sealed, and the mixture was stirred at room temperature for 48 hours. After the reaction mixture was filtered, the solid residue was additionally extracted with chloroform. All organics were combined, and the solvent was removed using a rotary evaporator. Then, the product was purified by chromatography (hexane–ethyl acetate as an eluent).One-pot reaction for the synthesis of 3-substituted isoxazoles 4 from aldoximes and calcium carbide
A reaction tube was loaded with an aldoxime (0.3 mmol), calcium carbide (2 mmol) and 0.4 mmol of N-chlorosuccinimide. Then, 1.5 ml of CCl4 was added, the resulting solution was stirred until the oxime dissolved, and then deionized water (4 mmol) was added. The reaction tube was immediately sealed, and the mixture was stirred at room temperature for 48 hours. After the reaction mixture was filtered, the solid residue was additionally washed with chloroform. All organics were combined, and the solvent was removed using a rotary evaporator. Then, the product was purified by chromatography (hexane–ethyl acetate as an eluent).General procedure for the synthesis of 4,5-dideuteroisoxazoles 5
All manipulations were carried out under an argon atmosphere. Anhydrous reagents and solvents were used. CDCl3 was treated with Al2O3 to avoid aldoxime E–Z isomerization in CDCl3 and was dried over molecular sieves to avoid D-H exchange with the water contaminants in chloroform.A reaction tube equipped with a magnetic stir bar was loaded with the aldoxime (100 mg), 0.75 ml of CDCl3 and 1.0 ml of D2O. The tube was sealed, and the mixture was stirred at 37 °C for 120 hours. Then, the organic layer was separated, and the solvent was evaporated. The resulting O-deuterated oxime was immediately used in the next step.
A reaction tube was loaded with D-aldoxime (0.25 mmol), calcium carbide (1.5 mmol) and 0.3 mmol of N-chlorosuccinimide. Then, 1.5 ml of CCl4 was added, the resulting solution was stirred until the oxime dissolved, and then D2O (8 mmol) was added. The reaction tube was immediately sealed, and the mixture was stirred at room temperature for 48 hours. After the reaction mixture was filtered, the solid residue was additionally washed with a small portion of chloroform. All organics were combined, and the solvent was removed using a rotary evaporator. Then, the product was purified by chromatography (hexane–ethyl acetate as an eluent).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the Russian Science Foundation (Project No 16-13-10301). This research made use of resources from the X-ray Diffraction Centre, Centre for Magnetic Resonance, Educational Resource Center of Chemistry and the Centre for Chemical Analysis and Materials of Saint-Petersburg State University.Notes and references
- (a) M. Shigenobu, K. Takenaka and H. Sasai, Angew. Chem., Int. Ed., 2015, 54, 9572 CrossRef CAS PubMed; (b) J.-H. Chu, C.-C. Chen and M.-J. Wu, Organometallics, 2008, 27, 5173 CrossRef CAS; (c) T. K. Goncharov, V. V. Dubikhin, E. L. Ignat'eva and G. M. Nazin, Russ. J. Phys. Chem. B, 2015, 9, 695 CrossRef CAS.
- M. Miyashita, M. Sasaki, I. Hattori, M. Sakai and K. Tanino, Science, 2004, 305, 495 CrossRef CAS PubMed.
- (a) D. J. Kushner, A. Baker and T. G. Dunstall, Can. J. Physiol. Pharmacol., 1999, 77, 79 CrossRef CAS PubMed; (b) S. L. Harbeson and R. D. Tung, Annu. Rep. Med. Chem., 2011, 46, 403 CAS; (c) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; (d) D. H. Phillips, G. A. Potter, M. N. Horton, A. Hewer, C. Crofton-Sleigh, M. Jarman and S. Venitt, Carcinogenesis, 1994, 15, 1487 CrossRef CAS PubMed; (e) M. Jarman, G. K. Poon, M. G. Rowlands, R. M. Grimshaw, M. N. Horton, G. A. Potter and R. McCague, Carcinogenesis, 1995, 16, 683 CrossRef CAS PubMed; (f) A. E. Mutlib, R. J. Gerson, P. C. Meunier, P. J. Haley, H. Chen, L. S. Gan, M. H. Davies, B. Gemzik and D. D. Christ, Toxicol. Appl. Pharmacol., 2000, 169, 102 CrossRef CAS PubMed.
- (a) A. Katsnelson, Nat. Med., 2013, 19, 656 CrossRef CAS PubMed; (b) F. Maltais, Y. C. Jung, M. Chen, J. Tanoury, R. B. Perni, N. Mani, L. Laitinen, H. Huang, S. Liao, H. Gao, D. F. Krahn, J. A. Markwalder, S. P. Seitz, R. T. Robertson and G. T. Miwa, J. Med. Chem., 2009, 52, 7993 CrossRef CAS PubMed; (c) A. Mullard, Nat. Rev. Drug Discovery, 2017, 16, 305 Search PubMed.
- The Chemistry of Heterocyclic Compounds, ed. A. Padwa and W. H. Pearson, 2002, vol. 59 Search PubMed.
- (a) A. J. Wilensky, P. N. Friel, L. M. Ojemann, C. B. Dodrill, K. B. McCormick and R. H. Levy, Epilepsia, 1985, 26, 212 CrossRef CAS PubMed; (b) H.-W. Chan, C.-Y. Huang, W.-J. Feng and Y.-C. Yen, J. Affective Disord., 2016, 205, 360 CrossRef CAS PubMed; (c) F. Fang, H. Sun, Z. Wang, M. Ren, J. R. Calabrese and K. Gao, CNS Drugs, 2016, 30, 845 CrossRef CAS PubMed; (d) A. Prakash and B. Jarvis, Drugs, 1999, 58, 1137 CrossRef CAS PubMed; (e) A. C. Bowen, S. Y. C. Tong, R. M. Andrews, I. M. O'Meara, M. I. McDonald, M. D. Chatfield, B. J. Currie and J. R. Carapetis, Lancet, 2014, 384, 2132 CrossRef CAS PubMed; (f) B. Negi, D. Kumar, W. Kumbukgolla, S. Jayaweera, P. Ponnan, R. Singh, S. Agarwal and D. S. Rawat, Eur. J. Med. Chem., 2016, 115, 426 CrossRef CAS PubMed; (g) K. A. M. Attia, M. W. I. Nassar, M. B. El-Zeiny and A. Serag, Spectrochim. Acta, Part A, 2016, 161, 64 CrossRef CAS PubMed; (h) J.-M. Dogné, C. T. Supuran and D. Pratico, J. Med. Chem., 2005, 48, 2251 CrossRef PubMed.
- (a) H. G. Leis, G. Fauler and W. Winischhofer, Curr. Org. Chem., 1998, 2, 131 CAS; (b) A. G. Burton, P. P. Forsythe, C. D. Johnson and A. R. Katritzky, J. Chem. Soc. B, 1971, 2365 RSC.
- J. H. Bowie, R. K. M. R. Kallury and R. G. Cooks, Aust. J. Chem., 1969, 22, 563 CrossRef CAS.
- O. Jackowski, T. Lecourt and L. Micouin, Org. Lett., 2011, 13, 5664 CrossRef CAS PubMed.
- (a) P. Allegretti and E. M. Ferreira, Chem. Sci., 2013, 4, 1053 RSC; (b) S. E. Lowe and J. Sheridan, Chem. Phys. Lett., 1978, 58, 79 CrossRef CAS.
- (a) K. S. Rodygin, G. Werner, F. A. Kucherov and V. P. Ananikov, Chem. – Asian J., 2016, 11, 965 CrossRef CAS PubMed; (b) R. Matake, Y. Adachia and H. Matsubara, Green Chem., 2016, 18, 2614 RSC; (c) E. Rattanangkool, T. Vilaivan, M. Sukwattanasinitt and S. Wacharasindhu, Eur. J. Org. Chem., 2016, 4347 CrossRef CAS; (d) K. S. Rodygin and V. P. Ananikov, Green Chem., 2016, 18, 482 RSC; (e) K. S. Rodygin, A. A. Kostin and V. P. Ananikov, Mendeleev Commun., 2015, 25, 415 CrossRef CAS; (f) G. Werner, K. S. Rodygin, A. A. Kostin, E. G. Gordeev, A. S. Kashin and V. P. Ananikov, Green Chem., 2017, 19, 3032 RSC; (g) A. Hosseini, D. Seidel, A. Miska and P. R. Schreiner, Org. Lett., 2015, 17, 2808 CrossRef CAS PubMed; (h) R. Matake, Y. Niwa and H. Matsubara, Org. Lett., 2015, 17, 2354 CrossRef CAS PubMed; (i) N. Kaewchangwat, R. Sukato, V. Vchirawongkwin, T. Vilaivan, M. Sukwattanasinitt and S. Wacharasindhu, Green Chem., 2015, 17, 460 RSC.
- (a) L. I. Belen'kii, Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis: Novel Strategies in Synthesis, ed. H. Feuer, Wiley, 2nd edn, 2008, pp. 1–128 Search PubMed; (b) F. Heaney, Eur. J. Org. Chem., 2012, 3043 CrossRef CAS; (c) L. Jiang, T. Gao, Z. Li, S. Sun, C. Kim, C. Huang, H. Guo, J. Wang and Y. Xing, Tetrahedron Lett., 2016, 57, 712 CrossRef CAS; (d) N. C. Tran, H. Dhondt, M. Flipo, B. Deprez and N. Willand, Tetrahedron Lett., 2015, 56, 4119 CrossRef CAS; (e) S. Grecian and V. V. Fokin, Angew. Chem., Int. Ed., 2008, 47, 8285 CrossRef CAS PubMed; (f) J. E. Grob, J. Nunez, M. A. Dechantsreiter and L. G. Hamann, J. Org. Chem., 2011, 76, 10241 CrossRef CAS PubMed.
- (a) M. J. Kohl and R. G. Lejeune, Steroids, 2002, 67, 71 CrossRef CAS PubMed; (b) O. V. Demina, A. A. Khodonov, E. I. Sinauridze, V. I. Shvets and S. D. Varfolomeeva, Russ. Chem. Bull., 2014, 63, 2092 CrossRef CAS.
- (a) P. Y. S. Lam, J. J. Adams, C. G. Clark, W. J. Calhoun, J. M. Luettgen, R. M. Knabb and R. R. Wexler, Bioorg. Med. Chem. Lett., 2003, 13, 1795 CrossRef CAS PubMed; (b) M. L. McIntosh, M. R. Naffziger, B. O. Ashburn, L. N. Zakharov and R. G. Carter, Org. Biomol. Chem., 2012, 10, 9204 RSC; (c) B. A. Chalyk, I. Y. Kandaurova, K. V. Hrebeniuk, O. V. Manoilenko, I. B. Kulik, R. T. Iminov, V. Kubyshkin, A. V. Tverdokhlebov, O. K. Ablialimov and P. K. Mykhailiuk, RSC Adv., 2016, 6, 25713 RSC; (d) M. A. Weidner-Wells, H. M. Werblood, R. Goldschmidt, K. Bush, B. D. Foleno, J. J. Hilliard, J. Melton, E. Wira and M. J. Macielag, Bioorg. Med. Chem. Lett., 2004, 14, 3069 CrossRef CAS PubMed.
- (a) P. Conti, A. Pinto, L. Tamborini, P. Dunkel, V. Gambaro, G. L. Visconti and C. De Micheli, Synthesis, 2009, 591 CrossRef CAS; (b) E. Coutouli-Argyropoulou, P. Lianis, M. Mitakou, A. Giannoulisa and J. Nowak, Tetrahedron, 2006, 62, 1494 CrossRef CAS.
- See section S1.2 in the ESI† for a detailed description of the experiments shown in Table 3.
- O. V. Demina, A. A. Khodonov, E. I. Sinauridze, V. I. Shvets and S. D. Varfolomeeva, Russ. Chem. Bull., 2014, 63, 2092 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1541266. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qo00705a |
| This journal is © the Partner Organisations 2018 |