
Catalytic asymmetric synthesis of pyrrolidine derivatives bearing heteroatom-substituted quaternary stereocenters†
Zhi-Peng
Wang
,
Qi
Wu
,
Jia
Jiang
,
Zi-Rui
Li
,
Xiao-Jiao
Peng
,
Pan-Lin
Shao
 * and
Yun
He
*
* and
Yun
He
*
Chongqing Key Laboratory of Natural Product Synthesis and Drug Research, School of Pharmaceutical Sciences, Chongqing University, 55 Daxuecheng South Road, Shapingba, Chongqing 401331, P.R. China. E-mail: shaopl@cqu.edu.cn; yun.he@cqu.edu.cn
First published on 18th September 2017
Abstract
An enantioselective [3 + 2] cycloaddition of heteroatom-substituted alkenes with α-substituted isocyanoacetates has been developed. The resulting cycloadducts could be further transformed to access a number of structurally diverse and biologically important pyrrolidine derivatives which bear sulfur- or oxygen-substituted quaternary stereocenters. Excellent reactivity and enantioselectivity were obtained for a broad range of substrates.
Introduction
Isocyanoacetates and their derivatives are widely used in organic, inorganic, coordination, polymeric, combinatorial, and medicinal chemistry.1 Moreover, their unique chemical reactivity renders them as versatile building blocks for the synthesis of biologically active molecules and natural products, and makes them very attractive for investigations.2 Our recent efforts have been devoted to using functionalized 1,3-dipole enolate derived from isocyanoacetate under chiral Brønsted base conditions.3 We have developed several asymmetric [3 + 2] cyclizations of isocyanoacetates with α-imino esters,3a allenoates,3b and methyleneindolinones3c with the aid of chiral silver(I) complexes developed by the Dixon group4 to synthesize oxazole-imidazolines, 3H pyrroles and spirooxindoles, respectively. While the nature of these reaction profiles necessitates further investigation, it provided more possibilities for our methodology to produce structurally diverse compounds for biological screening (Scheme 1).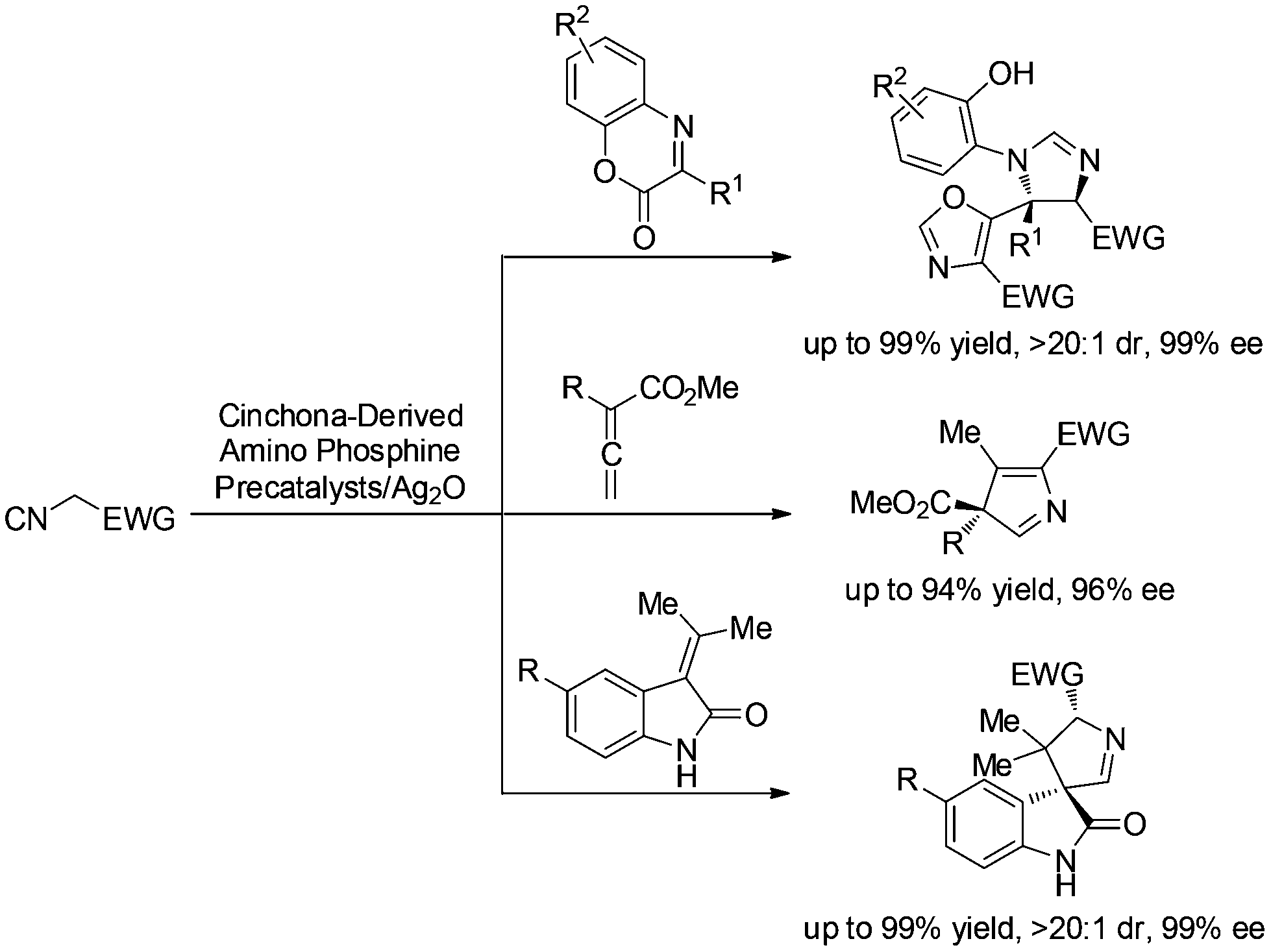 | ||
| Scheme 1 Representative [3 + 2] cycloaddition of isocyanoacetates. | ||
For several years, we have been pursuing the development of asymmetric synthesis of heterocyclic compounds, especially nitrogen-containing heterocycles,3,5 such as pyrrolidines and their analogues. Chiral pyrrolidine has been considered a “privileged scaffold” and exists in many biologically active natural products, organocatalysts and synthetic materials.6 In particular, pyrrolidine derivatives bearing heteroatom-substituted quaternary stereocenters may present more opportunities to these applications,7 especially for small-molecule drug design.8 Therefore, the development of catalytic enantioselective methods to construct such moieties would be highly desired. Herein we describe the asymmetric [3 + 2] cyclization of heteroatom-substituted alkenes9 with α-substituted isocyanoacetates to synthesize pyrrolidine derivatives bearing sulfur as well as oxygen substituted quaternary stereocenters via a sequential [3 + 2] cycloaddition and hydrogenation (Fig. 1).
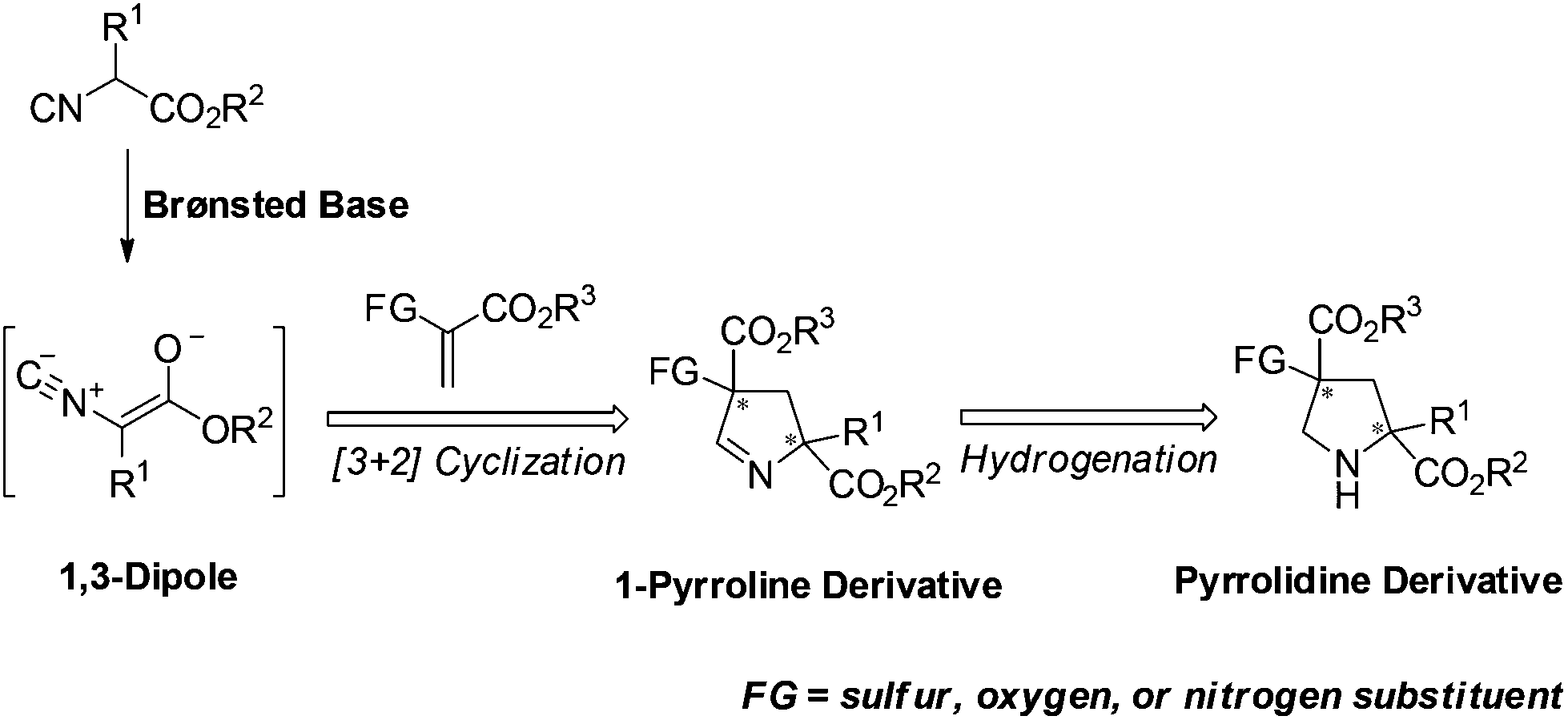 | ||
| Fig. 1 Catalytic asymmetric synthesis of pyrrolidines bearing heteroatom-substituted quaternary stereocenters. | ||
Results and discussion
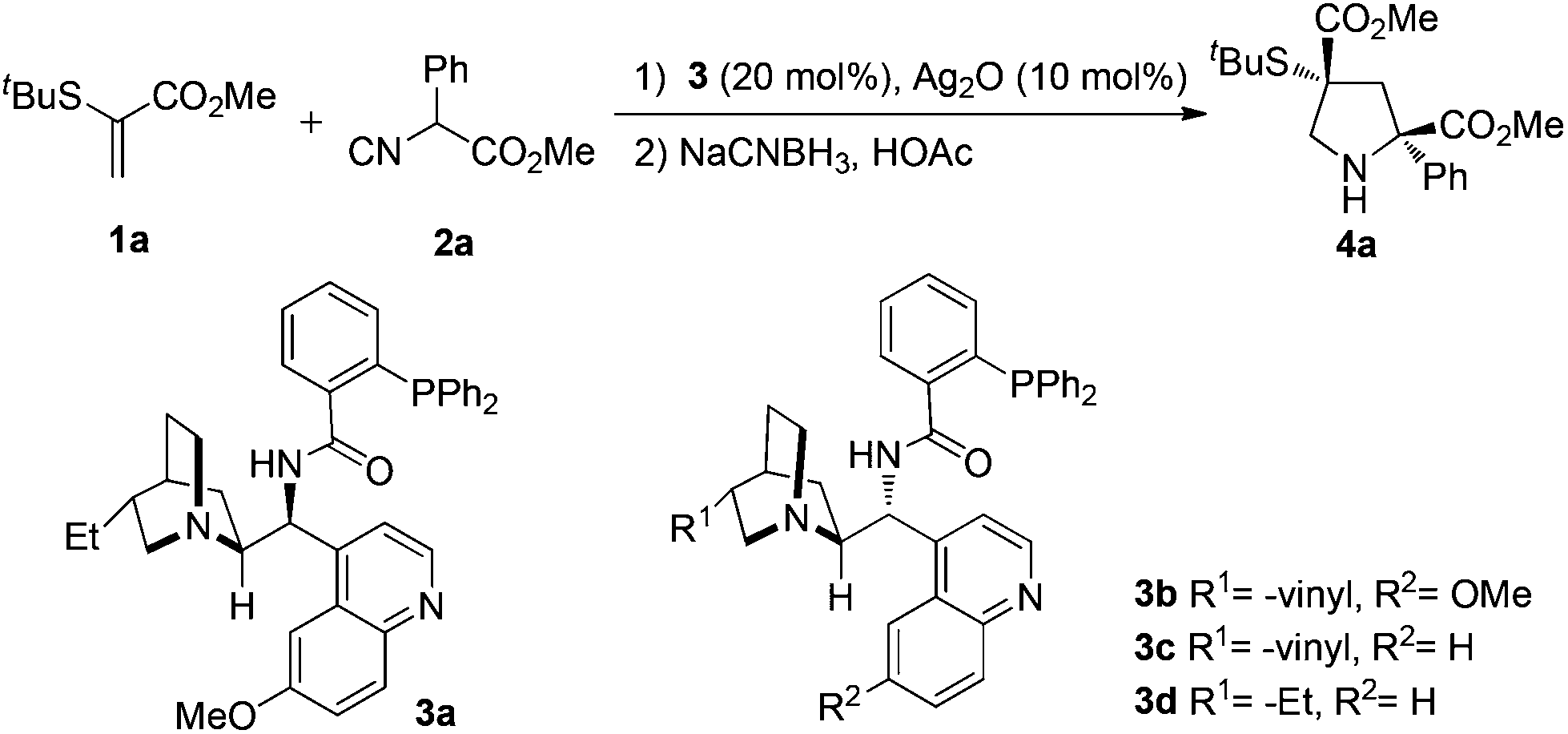
As shown in Table 1, the readily available methyl 2-(tert-butylthio)acrylate 1a and methyl 2-isocyano-2-phenylacetate 2a were chosen as the model substrates to investigate the feasibility of the [3 + 2] cycloaddition/reduction. Inspired by the recent reports from the Dixon group4 and based on our development on enantioselective isocyanoacetate cycloaddition catalyzed by silver(I) complexes,3 we tested the related cinchona-derived amino phosphine precatalysts for this process. To our great delight, the use of precatalyst 3a with Ag2O led to excellent yield (92%) and enantioselectivity (trans: 96% ee, cis: 93% ee) at ambient temperature, using THF as a solvent, albeit with poor diastereoselectivity (trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cis = 65:35) (entry 1). Lowering the temperature didn't significantly impact the cyclization (entries 5–8). A survey of solvents identified CHCl3 as the most suitable media (99% yield, trans:cis = 75:25, trans: 98% ee, cis: 96% ee) (entries 9–14). A series of optimizations were conducted, although higher diastereoselectivity was not obtained, the diastereomers bearing Cγ-tetrasubstituted α-thioether acid moieties could be easily separated by chromatography. Thus, we identified the following optimal protocol: the reaction of 1a and 2a with a molar ratio of 1.0:1.2 in the presence of Ag2O (10 mol%) and precatalyst 3a (20 mol%) in CHCl3 (1 mL) at −20 °C. It is worth noting that all the reactions were set up in open air without the need for exclusion of air or moisture.
:cis = 65:35) (entry 1). Lowering the temperature didn't significantly impact the cyclization (entries 5–8). A survey of solvents identified CHCl3 as the most suitable media (99% yield, trans:cis = 75:25, trans: 98% ee, cis: 96% ee) (entries 9–14). A series of optimizations were conducted, although higher diastereoselectivity was not obtained, the diastereomers bearing Cγ-tetrasubstituted α-thioether acid moieties could be easily separated by chromatography. Thus, we identified the following optimal protocol: the reaction of 1a and 2a with a molar ratio of 1.0:1.2 in the presence of Ag2O (10 mol%) and precatalyst 3a (20 mol%) in CHCl3 (1 mL) at −20 °C. It is worth noting that all the reactions were set up in open air without the need for exclusion of air or moisture.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Precatalyst | Solvent | Temp. | Yieldb [%] |
trans:cisc |
eed [%] (trans, cis) |
| a Reaction conditions: Ag2O (0.01 mmol), precatalyst 3 (0.02 mmol), 1a (0.10 mmol), 2a (0.12 mmol), solvent (1.0 mL), 24 h. b Total yields of the isolated trans and cis-isomer. c Determined by 1H NMR spectroscopy (400 MHz) of the reaction mixture. d Determined by HPLC analysis using a chiral stationary phase. See the ESI for details. | ||||||
| 1 | 3a | THF | 25 °C | 92 | 65:35 |
96, 93 |
| 2 | 3b | THF | 25 °C | 88 | 83:17 |
−95, −81 |
| 3 | 3c | THF | 25 °C | 91 | 84:16 |
−91, −76 |
| 4 | 3d | THF | 25 °C | 90 | 83:17 |
−92, −55 |
| 5 | 3a | THF | 0 °C | 93 | 66:34 |
97, 95 |
| 6 | 3a | THF | −10 °C | 93 | 68:32 |
98, 95 |
| 7 | 3a | THF | −20 °C | 95 | 68:32 |
98, 96 |
| 8 | 3a | THF | −40 °C | 95 | 69:31 |
95, 95 |
| 9 | 3a | CHCl3 | −20 °C | 99 | 75:25 |
98, 96 |
| 10 | 3a | DCM | −20 °C | 95 | 67:33 |
98, 97 |
| 11 | 3a | Toluene | −20 °C | 98 | 56:44 |
95, 95 |
| 12 | 3a | EA | −20 °C | 97 | 69:31 |
98, 95 |
| 13 | 3a | Et2O | −20 °C | 96 | 67:33 |
96, 95 |
| 14 | 3a | TBME | −20 °C | 95 | 68:32 |
93, 88 |
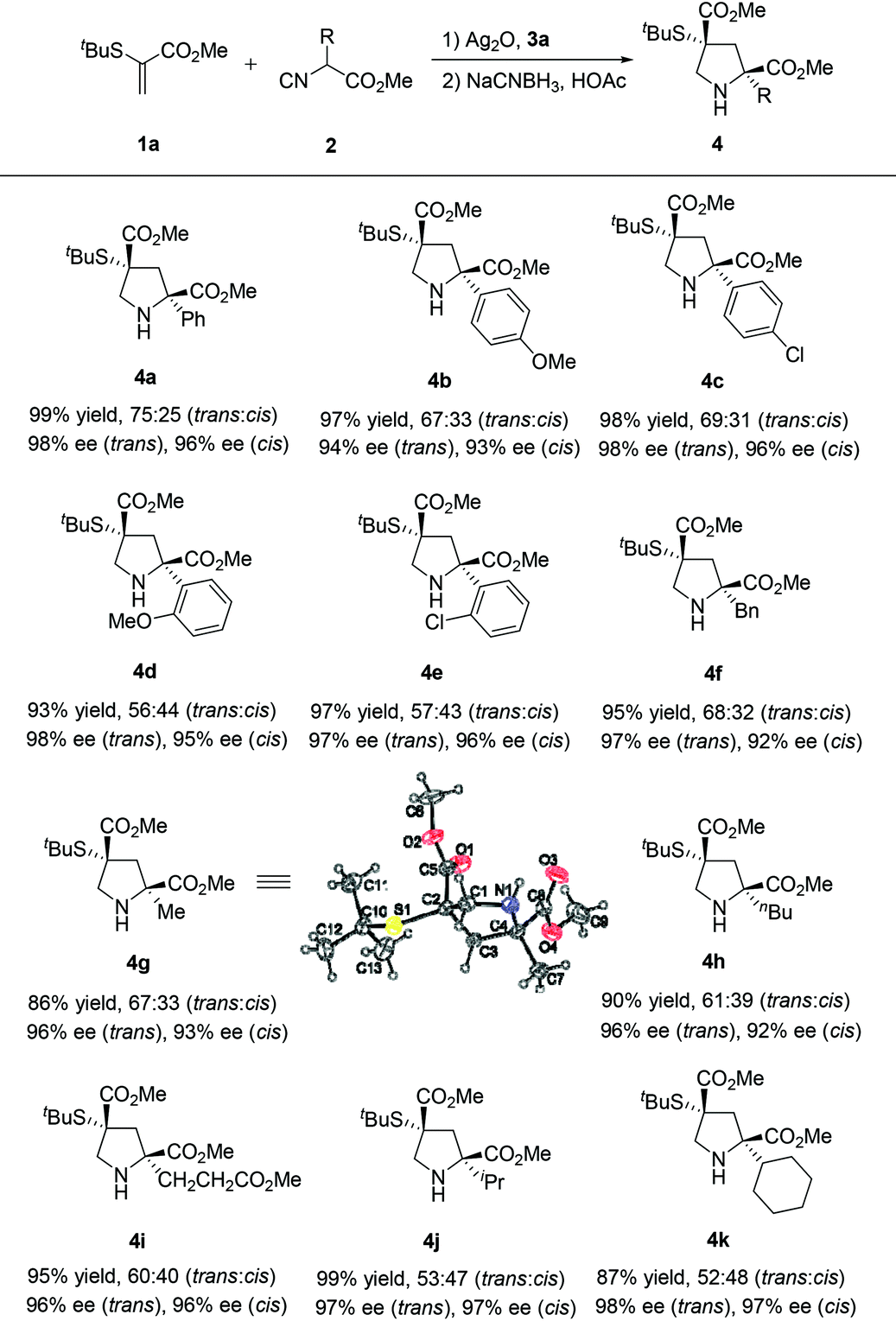
To study the utility of this catalytic regime, the effect of substituents in isocyanoacetates 2 with different acidity of the α-hydrogen atoms was investigated under the optimized conditions. To our delight, the [3 + 2] cyclization products 4 were obtained in uniformly high yields and enantioselectivities (Table 2). The substrates, bearing electron-neutral (4a), electron-rich (4b), electron-deficient (4c) and bulky (4d, 4e) aryl groups, participated in the reaction and proved to be suitable substrates, thus furnishing the corresponding products in excellent yields (86–99%) and enantioselectivities (trans: 94%–98% ee, cis: 92%–97% ee). Different substitution patterns on the isocyanoacetates, benzyl (4f), methyl (4g), butyl (4h), ester (4i), isopropyl (4j) and cyclohexyl (4k) groups were well tolerated. The relative and absolute configuration of 4g were established as (2S,4R) by single crystal X-ray analysis, and those of other products 4 were assigned by analogy.10 Notably, the formation of 4 highlighted the versatility of our method to prepare pyrrolidine derivatives possessing two quaternary stereocenters. This process represents an advancement of operational simplicity, synthetic efficiency and atom economy.
| a Unless otherwise noted, reactions were carried out with Ag2O (0.01 mmol), precatalyst 3a (0.02 mmol), 1a (0.10 mmol), 2 (0.12 mmol) in CHCl3 (1.0 mL) at −20 °C for 24 h. The dr values were determined by 1H NMR spectroscopy (400 MHz) of the reaction mixture. The yields given are those of the isolated of diastereoisomers. The enantiomeric excesses were determined by chiral HPLC analysis. See the ESI for details. |
|---|
|
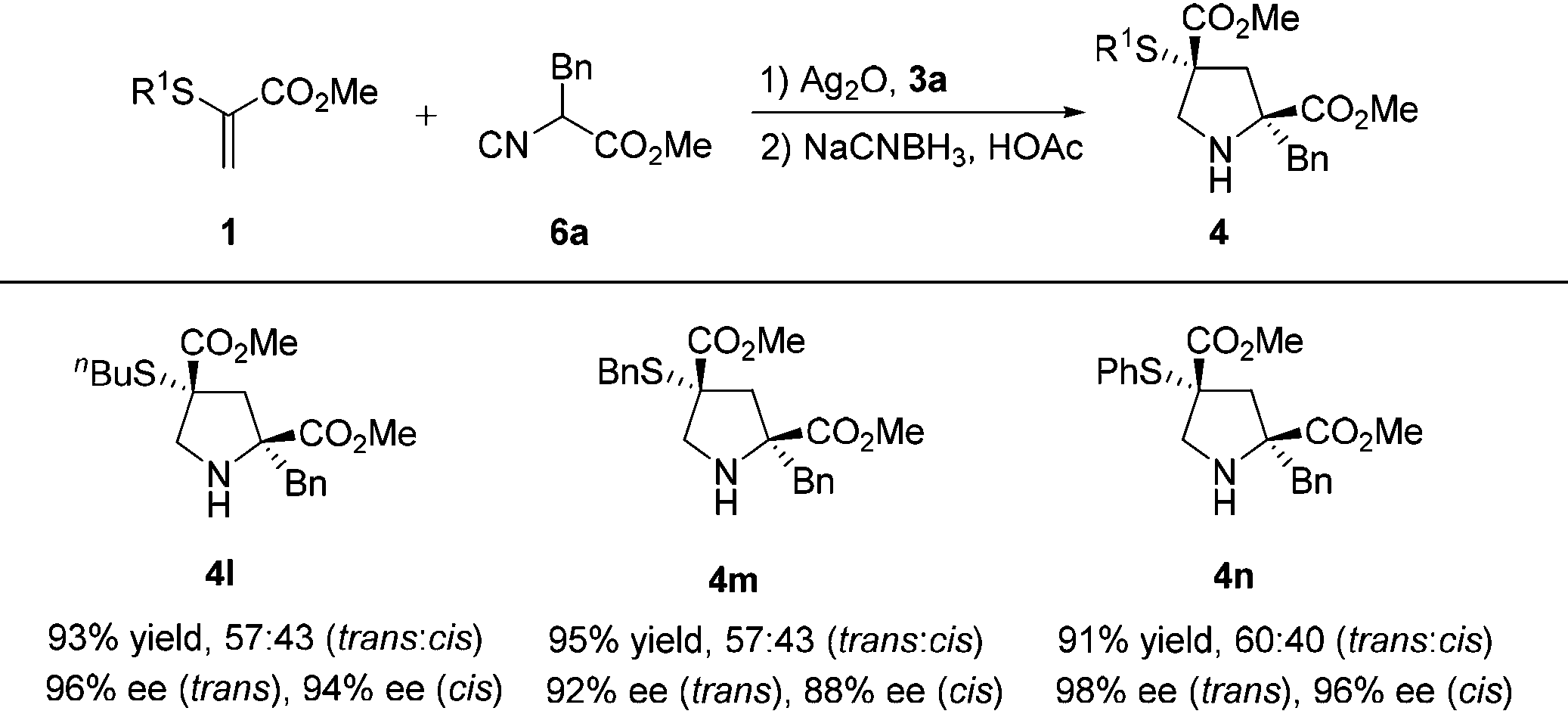
Using this catalytic protocol, α-thioacrylates 1 with alkyl or aryl groups were then examined for the [3 + 2] cyclization and the representative results are summarized in Table 3. These substituents at the α-thioacrylates (R1 = butyl, benzyl, phenyl) have no significant effect on the reactivity and selectivity. All the reactions went smoothly, providing products 4l–n in uniformly high yields (91%–93%) and excellent enantioselectivities (trans: 92%–98% ee, cis: 88%–96% ee).
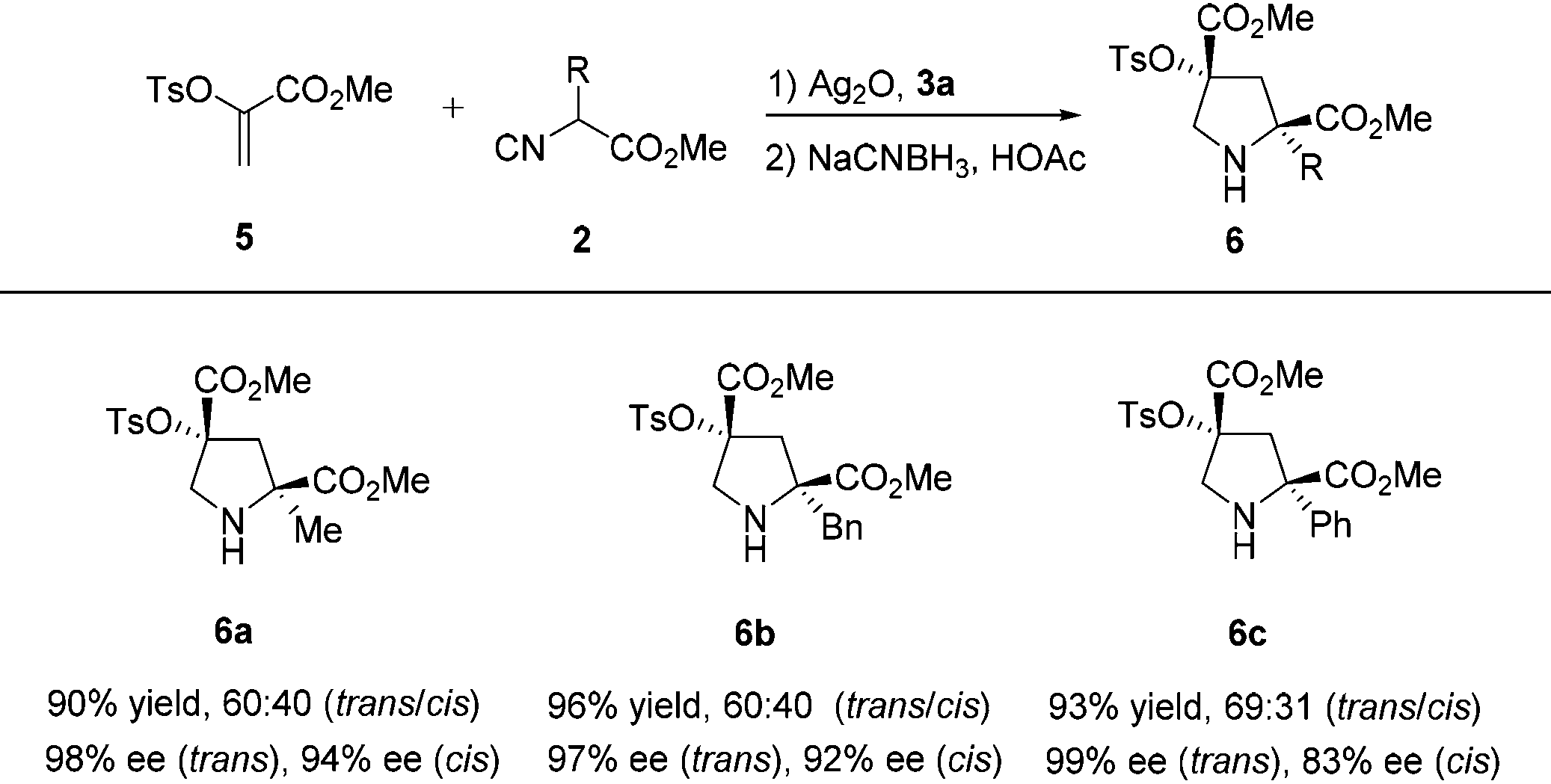
Then, we turned our attention to determine the scope of this process with other heteroatom substituents, with the goal to provide a versatile approach for drug leads. As expected, the same reaction conditions could be utilized for the sequential asymmetric [3 + 2] cycloaddition/reduction of α-tosyloxyacrylate 5 with α-substituted isocyanoacetates 2 to produce pyrrolidine derivatives 6, which bear Cγ-tetrasubstituted α-hydroxy acid moieties. Pyrrolidine derivatives with 2-methyl (6a), benzyl (6b), and phenyl (6c) groups were all produced in excellent yields (90%–96%) and enantioselectivities (trans: 97%–99% ee, cis: 83%–94% ee) from the corresponding α-substituted isocyanoacetates (Table 4).
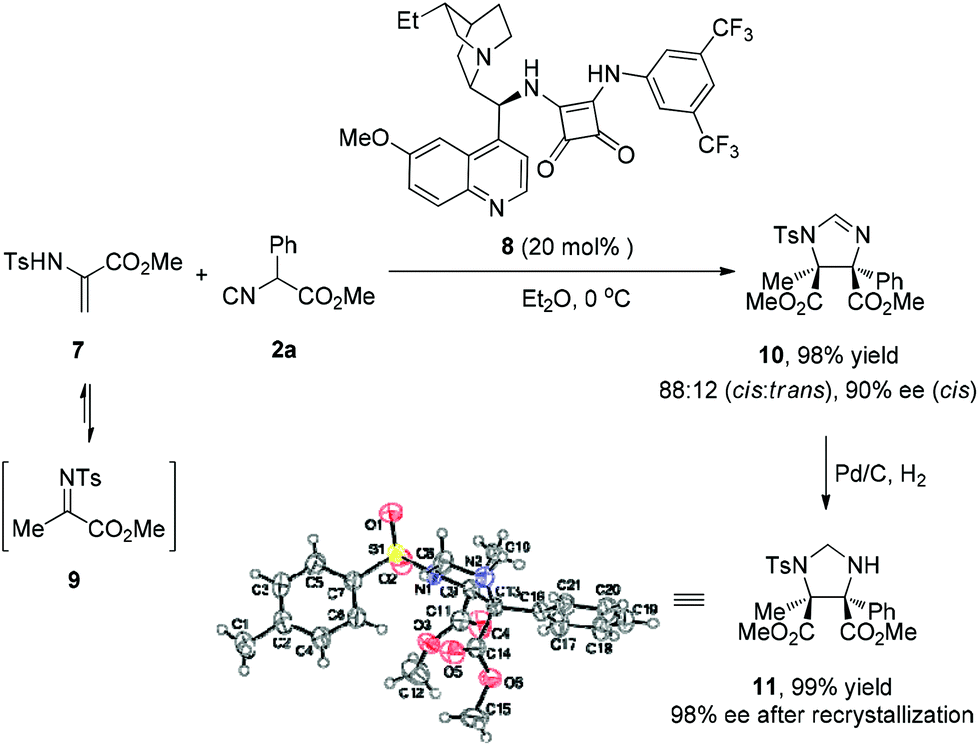
With respect to nitrogen-substituted alkenes, such as α,β-dehydroamino acids (7), which are reasonably reactive in Michael addition and cycloadditions,11 turned out to be difficult substrates using this catalytic system. Nevertheless, during the process employing the cinchona-derived bifunctional squaramide catalyst 8, methyl 2-(4-methylphenylsulfonamido)-acrylate 7 was transformed to methyl 2-(tosylimino)propanoate 9, which served as a ketimine in the reaction with isocyanoacetate 2a. This Mannich-type reaction proceeded smoothly, affording imidazoline 10 containing two quaternary stereocenters in excellent yield (98%) with high diastereoselectivity (cis:trans = 88:12) and enantioselectivity (90% ee) (Scheme 2). The imidazoline moiety could be reduced to yield the masked α,β-diamino ester 11, whose relative and absolute configuration were confirmed by X-ray analysis.10
 | ||
| Scheme 2 Organocatalytic [3 + 2] cyclization of 7 and 2a. See the ESI† for details. | ||
Conclusions
We have developed a catalytic enantioselective synthesis of the structurally diverse and biologically important pyrrolidine derivatives bearing two heteroatom-substituted quaternary stereocenters via a sequential [3 + 2] cycloaddition/reduction of α-substituted isocyanoacetates with heteroatom-substituted olefins, providing the corresponding cycloadducts in high yields (up to 99%) and excellent enantioselectivities (up to >99% ee). The high efficiency of this process, coupled with the operational simplicity, makes it an attractive method for pyrrole synthesis. Application of this protocol in drug discovery is ongoing in our laboratory.Experimental section
General procedure for the synthesis of pyrrolidine derivatives
To a 10 mL tube charged with 3a (12.3 mg, 0.02 mmol) and Ag2O (2.3 mg, 0.01 mmol) was added CHCl3 (1 mL). The mixture was stirred at −20 °C for 5 minutes, then α-thioacrylate 1a (17.4 mg, 0.10 mmol) and isocyanoacetate 2a (21.0 mg, 0.12 mmol) were added. The reaction mixture was stirred at −20 °C until 1a was consumed, and then filtered through a pad of silica gel and washed with ethyl acetate. The solvent was removed under reduced pressure and then the residue was dissolved in MeOH (1 mL), and then NaCNBH3 (12.6 mg, 0.20 mmol) and HOAc (12 mg, 0.20 mmol) were added sequentially at room temperature. The reaction mixture was stirred at room temperature for 0.5 h, concentrated and then purified by flash chromatography on silica gel (hexanes/ethyl acetate, 2:1) to afford product 4a.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the National Natural Science Foundation of China (no. 21402150, 21372267, 21572027) and Chongqing University for financial support. The authors thank Dr Yong-Liang Shao (Lanzhou University) and Xiangnan Gong (Chongqing University) for the X-ray crystallographic analysis.Notes and references
- (a) A. V. Gulevich, A. G. Zhdanko, R. V. A. Orru and V. G. Nenajdenko, Chem. Rev., 2010, 110, 5235 CrossRef CAS PubMed; (b) A. V. Lygin and A. Meijere, Angew. Chem., Int. Ed., 2010, 49, 9094 CrossRef CAS PubMed.
- (a) D. Zheng, S. Li and J. Wu, Org. Lett., 2012, 14, 2655 CrossRef CAS PubMed; (b) R. Ye, J. Zhao, B. Yuan, W.-C. Liu, J. R. D. Araujo, F. F. Faucher, M. Chang, C. V. Deraedt, F. D. Toste and G. A. Somorjai, Nano Lett., 2017, 17, 584 CrossRef CAS PubMed; (c) S. Qiao, C. B. Wilcox, D. K. Unruh, B. Jiang and G. Li, J. Org. Chem., 2017, 82, 2992 CrossRef CAS PubMed; (d) D. Zheng, S. Li, Y. Luo and J. Wu, Org. Lett., 2011, 13, 6402 CrossRef CAS PubMed; (e) H. Zhou, W. Zhang and B. Yan, J. Comb. Chem., 2010, 12, 206 CrossRef CAS PubMed; (f) X. Xu, L. Zhang, X. Liu, L. Pan and Q. Liu, Angew. Chem., Int. Ed., 2013, 52, 9271 CrossRef CAS PubMed; (g) T. Buyck, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2013, 52, 12714 CrossRef CAS PubMed; (h) M. Hayashi, M. Iwanaga, N. Shiomi, D. Nakane, H. Masuda and S. Nakamura, Angew. Chem., Int. Ed., 2014, 53, 8411 CrossRef CAS PubMed; (i) J. Liu, Z. Fang, Q. Zhang, Q. Liu and X. Bi, Angew. Chem., Int. Ed., 2013, 52, 6953 CrossRef CAS PubMed; (j) J. Tan, X. Xu, L. Zhang, Y. Li and Q. Liu, Angew. Chem., Int. Ed., 2009, 48, 2868 CrossRef CAS PubMed; (k) D. K. Tiwari, J. Pogula, B. Sridhar, D. K. Tiwari and P. R. Likhar, Chem. Commun., 2015, 51, 13646 RSC; (l) J.-Y. Liao, Q. Ni and Y. Zhao, Org. Lett., 2017, 19, 4074 CrossRef CAS PubMed; (m) J.-Y. Liao, W. J. Yap, J. Wu, M. W. Wong and Y. Zhao, Chem. Commun., 2017, 53, 9067 RSC.
- (a) P.-L. Shao, J.-Y. Liao, Y. A. Ho and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 5435 CrossRef CAS PubMed; (b) J.-Y. Liao, P.-L. Shao and Y. Zhao, J. Am. Chem. Soc., 2015, 137, 628 CrossRef CAS PubMed; (c) X.-J. Peng, Y. A. Ho, Z.-P. Wang, P.-L. Shao, Y. Zhao and Y. He, Org. Chem. Front., 2017, 4, 81 RSC.
- (a) R. de la Campa, I. Ortín and D. J. Dixon, Angew. Chem., Int. Ed., 2015, 54, 4895 CrossRef CAS PubMed; (b) I. Ortín and D. J. Dixon, Angew. Chem., Int. Ed., 2014, 53, 3462 CrossRef PubMed; (c) F. Sladojevich, A. Trabocchi, A. Guarna and D. J. Dixon, J. Am. Chem. Soc., 2011, 133, 1710 CrossRef CAS PubMed; (d) A. Franchino, P. Jakubec and D. J. Dixon, Org. Biomol. Chem., 2016, 14, 93 RSC.
- (a) P.-L. Shao, X.-Y. Chen and S. Ye, Angew. Chem., Int. Ed., 2010, 49, 8412 CrossRef CAS PubMed; (b) P.-L. Shao, X.-Y. Chen, L.-H. Sun and S. Ye, Tetrahedron Lett., 2010, 51, 2316 CrossRef CAS; (c) P. Shao, L. Shen and S. Ye, Chin. J. Chem., 2012, 30, 2688 CAS; (d) Y.-R. Zhang, L. He, X. Wu, P.-L. Shao and S. Ye, Org. Lett., 2008, 10, 277 CrossRef CAS PubMed; (e) X.-L. Huang, L. He, P.-L. Shao and S. Ye, Angew. Chem., Int. Ed., 2009, 48, 192 CrossRef CAS PubMed; (f) X.-N. Wang, P.-L. Shao, H. Lv and S. Ye, Org. Lett., 2009, 11, 4029 CrossRef CAS PubMed; (g) L.-T. Shen, P.-L. Shao and S. Ye, Adv. Synth. Catal., 2011, 353, 1943 CrossRef CAS.
- (a) V. H. Lauridsen, L. Ibsen, J. Blom and K. A. Jørgensen, Chem. – Eur. J., 2016, 22, 3259 CrossRef CAS PubMed; (b) H. Wang, Q. Deng, Z. Zhou, S. Hu, Z. Liu and L.-Y. Zhou, Org. Lett., 2016, 18, 404 CrossRef CAS PubMed; (c) A. L. Gerten and L. M. Stanley, Org. Chem. Front., 2016, 3, 339 RSC; (d) D.-J. Zhang, M.-S. Xie, G.-R. Qu, Y.-W. Gao and H.-M. Guo, Org. Lett., 2016, 18, 820 CrossRef CAS PubMed; (e) L.-W. Tang, B.-J. Zhao, L. Dai, M. Zhang and Z.-M. Zhou, Chem. – Asian J., 2016, 11, 2470 CrossRef CAS PubMed; (f) A. Cayuelas, R. Ortiz, C. Nájera, J. M. Sansano, O. Larrañga, A. de Cózar and F. P. CossÍo, Org. Lett., 2016, 18, 2926 CrossRef CAS PubMed; (g) A. Ponce, I. Alonso, J. Adrio and J. C. Carretero, Chem. – Eur. J., 2016, 22, 4952 CrossRef CAS PubMed; (h) J.-Y. Li, H. Kim, Y. Hun and K. Oh, Adv. Synth. Catal., 2016, 358, 984 CrossRef CAS; (i) Z.-M. Zhang, B. Xu, S. Xu, H.-H. Wu and J. Zhang, Angew. Chem., Int. Ed., 2016, 55, 6324 CrossRef CAS PubMed; (j) T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366 CrossRef CAS PubMed; (k) J. Adrio and J. C. Carretero, Chem. Commun., 2014, 50, 12434 RSC; (l) C. Nájera and J. M. Sansano, J. Organomet. Chem., 2014, 771, 78 Search PubMed; (m) B. Xu, Z.-M. Zhang, S. Xu, B. Liu, Y. Xiao and J. Zhang, ACS Catal., 2017, 7, 210 CrossRef CAS; (n) L. M. Stanley and M. P. Sibi, Chem. Rev., 2008, 108, 2887 CrossRef CAS PubMed; (o) M. Kuhnert, A. Blum, H. Steuber and W. E. Diederich, J. Med. Chem., 2015, 58, 4845 CrossRef CAS PubMed.
- (a) M. U. Luescher and J. W. Bode, Org. Lett., 2016, 18, 2652 CrossRef CAS PubMed; (b) S. Schann, C. Menet, P. Arvault, G. Mercier, M. Frauli, S. Mayer, N. Hubert, N. Triballeau, H.-O. Bertrand, F. Acher and P. Neuville, Bioorg. Med. Chem. Lett., 2006, 16, 4856 CrossRef CAS PubMed; (c) L. Tian, G.-Q. Xu, Y.-H. Li, Y.-M. Liang and P.-F. Xu, Chem. Commun., 2014, 50, 2428 RSC; (d) Q. Zhao and C. E. Schafmeister, J. Org. Chem., 2015, 80, 8968 CrossRef CAS PubMed; (e) M. González-Esguevillas, J. Adrio and J. C. Carretero, Chem. Commun., 2013, 49, 4649 RSC; (f) C. G. Levins and C. E. Schafmeister, J. Am. Chem. Soc., 2003, 125, 4702 CrossRef CAS PubMed; (g) M. Kheirabadi, N. Çelebi-Ölçüm, M. F. L. Parker, Q. Zhao, G. Kiss, K. N. Houk and C. E. Schafmeister, J. Am. Chem. Soc., 2012, 134, 18345 CrossRef CAS PubMed; (h) Z. Z. Brown and C. E. Schafmeister, Org. Lett., 2010, 12, 1436 CrossRef CAS PubMed; (i) J. L. G. Ruano, A. Fraile, M. R. Martín, G. González, C. Fajardo and A. M. Martín-Castro, J. Org. Chem., 2011, 76, 3296 CrossRef PubMed; (j) Q. Zhao, Y.-h. Lam, M. Kheirabadi, C. Xu, K. N. Houk and C. E. Schafmeister, J. Org. Chem., 2012, 77, 4784 CrossRef CAS PubMed; (k) S. Wu, N. Liu, G. Dong, L. Ma, S. Wang, W. Shi, K. Fang, S. Chen, J. Li, W. Zhang, C. Sheng and W. Wang, Chem. Commun., 2016, 52, 9593 RSC; (l) Y. Gui, L. Qiu, Y. Li, H. Li and S. Dong, J. Am. Chem. Soc., 2016, 138, 4890 CrossRef CAS PubMed; (m) K. Uchida, T. Ogawa, Y. Yasuda, H. Mimura, T. Fujimoto, T. Fukuyama, T. Wakimoto, T. Asakawa, Y. Hamashima and T. Kan, Angew. Chem., Int. Ed., 2012, 51, 12850 CrossRef CAS PubMed; (n) T. H. Lambert and S. J. Danishefsky, J. Am. Chem. Soc., 2006, 128, 426 CrossRef CAS PubMed; (o) S. W. B. Tan, C. L. L. Chai, M. G. Moloney and A. L. Thompson, J. Org. Chem., 2015, 80, 2661 CrossRef CAS PubMed; (p) A. X. Wang and P. M. Scola, WO2009146347, 2009 Search PubMed; (q) P. Gupta, J. Wan and S. T. Trzaska, WO2016100147, 2016 Search PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- Y. Fujiwara and G. C. Fu, J. Am. Chem. Soc., 2011, 133, 12293 CrossRef CAS PubMed.
- CCDC 1538775 (4g) and 1538777 (11) contain the supplementary crystallographic data for this paper. Selected data are also included in the ESI.†.
- (a) Z. Wang, S. Luo, S. Zhang, W.-L. Yang, Y.-Z. Liu, H. Li, X. Luo and W.-P. Deng, Chem. – Eur. J., 2013, 19, 6739 CrossRef CAS PubMed; (b) Z. Wang, X. Yu, B. X. Tian, D. T. Payne, W.-L. Yang, Y.-Z. Liu, J. S. Fossey and W.-P. Deng, Chem. – Eur. J., 2015, 21, 10457 CrossRef CAS PubMed; (c) W.-L. Yang, F.-F. Tang, F.-S. He, C.-Y. Li, X. Yu and W.-P. Deng, Org. Lett., 2015, 17, 4822 CrossRef CAS PubMed; (d) C. Yang, E.-G. Zhang, X. Li and J.-P. Cheng, Angew. Chem., Int. Ed., 2016, 55, 6506 CrossRef CAS PubMed; (e) K. F. W. Hekking, D. C. J. Waalboer, M. A. H. Moelands, F. L. van Delft and F. P. J. T. Rutjes, Adv. Synth. Catal., 2008, 350, 95 CrossRef CAS; (f) J. Y. L. Chung, M. Shevlin, A. Klapars and M. Journet, Org. Lett., 2016, 18, 1812 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, copies of 1H and 13C NMR spectra. CCDC 1538775 (4g) and 1538777 (11). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qo00692f |
| This journal is © the Partner Organisations 2018 |