
Designing a main-chain visible-light-labile picolinium-caged polymer and its biological applications†
Tongtong
Zhou
a,
Tao
Liu
b,
Yu
Bao
c,
Ping
Zhang
a,
Casey
Yan
a,
Fujun
Yao
a,
Shuxun
Cui
 c,
Yongming
Chen
d,
Xin
Chen
*b and
You
Yu
*a
c,
Yongming
Chen
d,
Xin
Chen
*b and
You
Yu
*a
aKey Laboratory of Synthetic and Natural Functional Molecular Chemistry of the Ministry of Education, College of Chemistry and Materials Science, Northwest University, Xi'an 710069, China. E-mail: yuyou@nwu.edu.cn
bSchool of Chemical Engineering and Technology, Institute of Polymer Science in Chemical Engineering, Xi'an Jiao Tong University, Xi'an 710049, China. E-mail: chenx2015@xjtu.edu.cn
cKey Laboratory of Advanced Technologies of Materials of Ministry of Education, Southwest Jiaotong University, Chengdu 610031, China
dSchool of Chemistry and Chemical Engineering, Key Laboratory for Polymeric Composite and Functional Materials of Ministry of Education, Sun Yat-Sen University, Guangzhou 510275, China
First published on 1st December 2017
Abstract
Designing a main-chain visible-light-labile polymer is still a challenging task in related fields. In this work, a picolinium-caged main-chain photolabile polymer (MCPP) is firstly reported, where the C–O bonds of picolinium groups are efficiently cleaved (>95%) upon visible light irradiation (452 nm). The degradation rate of the MCPP is easily tuned by adjusting the concentration of the catalyst, irradiation intensity and time. Importantly, after photodegradation, the molecular weight of degradation products is decreased to less than 650 g mol−1. It is found that, unlike forming a helix structure of pure polyethylene glycol in water, the picolinium-caged MCPP is in a linear conformation. Moreover, nanopore-based single-molecule analysis indicates that both the MCPP and degradation products can easily pass through a biomimetic lipid membrane, and the passing speed can be tuned by varying the applied bias voltage. Cell cytotoxicity and proliferation experiments demonstrate that both the MCPP and degradation products are nontoxic, but only the MCPP is favorable for cell proliferation. On the basis of these results, it is anticipated that the MCPP is an exceptional candidate of photodegradable materials in chemistry, pharmacy, materials science and medicine.
Introduction
Photolabile (or photodegradable) polymers have attracted considerable attention because they allow the alteration of polymer properties simply by light irradiation. To date, numerous amounts of main- and/or side-chain photolabile polymers are designed, showing exceptionally potential applications in the fields of biological and materials sciences.1–5 Compared with other stimuli (pH, temperature, enzymes and chemicals), light is undoubtedly a clean and readily available energy source to cleave photolabile bonds via a noncontact spatiotemporal manner.6–9 For example, UV light and near-infrared light are widely adopted to trigger the cleavage of photolabile nitrobenzyl derivatives,10–12 aliphatic ketones,13 coumarins14,15 and others.16–19 However, it is worth noting that light at longer wavelengths (>400 nm) is a safer stimulus when compared to these undesirable high-energy light sources (UV light or γ ray) where potential hazards to human bodies and active substances are inevitable. Furthermore, previous work indicates that the labile units caged in polymer backbones are helpful for their rapid degradation behaviors.2N-Alkyl Picolinium Esters (NPEs) are kinds of visible-light-labile groups that can be efficiently photocleaved via a photo-induced electron transfer process with electron mediators,20–23 especially the combination of the ruthenium complex (Ru(II))/ascorbic acid (ASC).20,24,25 In principle, upon visible light irradiation (∼452 nm), the excited Ru(II) is firstly reduced by ASC, and then returns to its normal state by oxidizing NPEs nearby. As a consequence, C–O bonds of NPEs are cleaved, simultaneously releasing negative acids and positive picoliniums. Taking the advantages of their rapid C–O bond fragmentation and charge conversion, NPEs have been used for releasing small molecules, preparing polymers, and tuning functions of materials.21,23–28 For instance, Zhu et al. designed a photo-controlled release system in the presence of (CdSe/ZnS) dots/ASC, which was available for photoreleasing 5-fluorouracil acid, an anticancer drug from the NPE ester.21 Falvey et al. used cinnamic acid-contained NPEs to orthogonally control aqueous solution viscosity via the photocleavage and transformation of NPEs.26 Recently, we reported two typical NPE-based materials for cargo delivery and the assembly/disassembly of multilayer films, which were based on the aforementioned photo-induced charge conversion of NPE units.24,25 In normal states, cargos and assembled films were stable in substrates, while both of them were released or erased from substrates upon visible light irradiation. However, as far as we are concerned, there is a rare report on applying NPEs in main-chain visible-light-labile polymers that show applications such as fast degradable materials, “burst” release of drug molecules and so on.
To address this issue, we report herein a main-chain photolabile polymer (MCPP) via a step-growth polymerization of modified HO–(CH2–CH2–)3–OH monomers, where the visible light photolabile picoliniums are caged into the main chains as the degradable units. The as-synthesized MCPP can be efficiently degraded (∼95%) with the catalysis of Ru(II)/ASC upon blue light irradiation. The degradation rate is easily controlled by adjusting the concentration of the catalyst, irradiation intensity and time. After photodegradation, the molecular weight of degradation products decreases to less than 650 g mol−1. It is found that, unlike forming a helix structure of polyethylene glycol in water, the picolinium-caged MCPP is in a linear conformation. Moreover, nanopore-based single-molecule analysis indicates that both the MCPP and degradation products can easily pass through biomimetic cell membranes, and the passing speed can be tuned by varying the applied bias voltage. Cell cytotoxicity and proliferation experiments demonstrate that both the MCPP and degradation products are nontoxic, but only the MCPP is favorable for cell proliferation. On the basis of these results, it is anticipated that the MCPP is an exceptional candidate of photodegradable materials in chemistry, pharmacy, materials science and medicine.
Results and discussion
As shown in Fig. 1, to prepare such a visible-light-labile MCPP, two typical monomers (i.e., 1 and 2) were firstly synthesized by functionalizing HO–(CH2–CH2–O)4–H with 4-pyridylcarbinol and iodine, respectively. The MCPP was then obtained by the step-growth polymerization of 1 and 2 in solvents, generating one picolinium unit between two –O–(CH2–CH2–O)3– chains via the highly efficient N-alkylation reaction of pyridine in 1 and iodine in 2 (Fig. S1 and S2†). After this, in order to realize the degradation behavior, the as-prepared MCPP, Ru(II) and ascorbic acid (ASC) were mixed together in water and exposed to blue light for the predetermined time, where the C–O bond of 4-picolinium were rapidly cleaved via a photo-induced electron transfer process. As a consequence, the MCPP was photodegraded into small fragments of 3 and 4. To understand well the properties of the MCPP, commercial polyethylene glycol (PEG) was used as the reference, because of the partially similar structure between them. | ||
| Fig. 1 The synthesis and visible light-triggered degradation of MCPP. (i) 4-Pyridylcarbinol, EDC, DMAP, and CH2Cl2; (ii) pTsCl, THF, and NaOH; (iii) NaI and acetone; (iv) CH3CN and reflux; (v) and ascorbic acid, [Ru(bpy)3]Cl2 and blue light (452 nm). | ||
The glass transition (Tg) and degradation (Td) temperatures of the as-prepared MCPP were about 3.4 °C and 255 °C, respectively, and the Td was higher than that of pure PEG2k (∼170 °C, Tg was not detected in the range from −50 to 40 °C) because of the rigid picolinium being introduced into the flexible main chains (Fig. S3a and S3b†). The cyclic voltammetry curve in Fig. S3c† shows that the redox potential of this polymer was about −0.97 V. Accordingly, the corresponding Gibbs free energy (ΔG) for the possible direct electron transfer (DET) and mediated electron transfer (MET) processes were 3.2 and −8.3 kcal mol−1, respectively, indicating a MET pathway for the C–O cleavage of NPEs in an aqueous solution of Ru(II)/ASC.24 Furthermore, by varying solvents and concentrations of monomers for the condensation polymerization, it was found that the maximum intrinsic viscosity ([η]) was 8.5 ml g−1 (Table S1†), when the MCPP was prepared at a concentration of 0.2 g mL−1 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 = 1:1.02, molar ratio) in acetonitrile. Thus, if not specially mentioned, all of the MCPP used later was synthesized under this optimized condition.
:2 = 1:1.02, molar ratio) in acetonitrile. Thus, if not specially mentioned, all of the MCPP used later was synthesized under this optimized condition.
To confirm the photodegradation behavior of the MCPP, MALDI-TOF-MS and GPC measurements were used to verify the molecular weight of samples before and after photodegradation. However, there was no significant difference observed in the GPC measurement, and even no signal was detected when water was used as the eluent (Fig. S4†). The reason for this was assigned to the strong absorption of the pyridinium-based MCPP to certain columns.29 For MALDI-TOF-MS, it is a powerful tool for characterizing charged polymers, especially with low molecular weight. As shown in Fig. 2a, the as-prepared MCPP had the molecular weight ranging from 1.5 to 2.5 kDa, where the repeat unit (ca. 44 Da) was assigned to the structure of –O–CH2–CH2–. After irradiation, the molecular weight was sharply decreased to less than 650 Da (3: 222 g mol−1, 4: 600 g mol−1), revealing an efficient photodegradation of the MCPP by the catalysis of Ru(II) and ASC as expected.
 | ||
| Fig. 2 (a) The MALDI-TOF-MS and (b) [η] of MCPP before (top) and after (bottom) photodegradation. (c) The chemical structures of MCPP and the picolinium-contained photodegraded product (5) after ion-exchanging. (d and e) The corresponding 1H NMR spectra of MCPP in D2O and 5 in CD3CN, respectively. The symbols star and triangle indicate the solvent peak and unidentified product, respectively. The photodegrading condition: CASC: 0.62 mol L−1, CRu: 45 μmol L−1, nASC/nC–O: 10, I: 30 mW cm−2, t: 12 h. | ||
Considering that intrinsic viscosity ([η]) is proportionally related to the molecular weight of polymers, the photodegradation process of the MCPP can be traced by measuring the change in [η]. Fig. 2b shows that [η] decreased from 8.5 to 4.9 ml g−1 after irradiation, which strongly supported the degradation behavior observed in Fig. 1 and 2a. Furthermore, the NMR characterization of the degradation product was employed to identify the photodegradation of the MCPP. To avoid the interference of Ru(II)/ASC, the hydrophobic 4-picolinium-ended molecules (5) were isolated in advance from the reaction mixture of 3, 4 and Ru(II)/ASC by an ion-exchange-induced precipitation (I− → Tf2N−, Fig. 2c). The comparison of the 1H-NMR spectra of MCPP and 5 showed that the peak at δ 5.4–5.5 (from picolinium) almost disappeared after irradiation (Fig. 2d and e). This difference could be ascribed to the cleavage of C–O bonds within NPE units. All of these characterization techniques indicated that the MCPP was successfully synthesized by caging photolabile picolinium into polymer backbones, and featured good photodegradation properties.
Inspired by the difference of NMR spectra before and after photodegradation as shown in Fig. 2d and e, 1H-NMR characterization was used to systematically evaluate the effect of different reaction conditions on the degradation efficiency (DE%): r was defined as the ratio of integrated areas of the peaks between δ 5.4–5.5 and δ 8.4–8.8. Thus, the degradation efficiency was easily calculated by using DE% = (1 − r) × 100%. Fig. 3a shows the 1H-NMR spectra of the degradation products at different molar ratios of ASC used and C–O bonds (nASC/nC–O) in NPEs, where nC–O was calculated from 1 used in synthesizing the MCPP (shown in Fig. 1). It was found that by increasing the nASC/nC–O, DE% increased gradually from 0 to >95% (Fig. 3b and entries 1–4 in Table S2†). Besides, the irradiation time had a significant influence on DE% which increased with prolonged irradiation time from 0.3 h to 12 h (Fig. 3c and entries 4–7 in Table S2†). However, the comparison studies indicated that both the concentrations of Ru(II) and irradiation intensity had little influence on DE% (Fig. 3d, entries 4, 8 and 9 in Table S2†). When increasing the concentration of Ru(II) tenfold, DE% only increased from 85% to 93%. And increasing the irradiation intensity twice resulted in the increase of DE% from 78% to 93%. Based on these results, it can be predicted that DE% was mainly determined by the concentration of ASC and the irradiation time. This prediction was in agreement with the reported results.20,24,25 Adding more ASC and increasing the irradiation time were beneficial for the conversion of Ru(II) to Ru(I) to catalyze the cleavage reaction of C–O bonds in picolinium units (Fig. 1, 3b and c).
 | ||
| Fig. 3 (a) 1H NMR spectra of the degradation products with different molar ratios of ASC and C–O bonds (nASC/nC–O) within picolinium units (t: 12 h, I: 30 mW cm−2, CRu(II): 45 μmol L−1). (b) The DE% calculated from the NMR results in a. Insets show the equations for calculating the ratio (r) of integrated areas of the peaks between δ 5.4–5.5 and δ 8.4–8.8, and the degradation efficiency (DE%) of MCPP. (c) The DE% of MCPP with different irradiation times (CASC: 0.62 mol L−1, CRu(II): 45 μmol L−1, nASC/nC–O: 10, I: 30 mW cm−2). (d) The DE% of MCPP with different concentrations of Ru(II) (CASC: 0.62 mol L−1, nASC/nC–O: 10, I: 30 mW cm−2, t: 12 h) and light intensities (CASC: 0.62 mol L−1, nASC/nC–O: 10, CRu(II): 45 μmol L−1, t: 12 h). | ||
Polymer conformation can dramatically change the properties of polymers and affect many binding reactions of the polymer with other substances. For example, in PEG–protein complexes, PEG can induce the conformational change of proteins and vary the protein–PEG interactions.30 Previous studies demonstrated that PEG did not aggregate in water but formed a supra-structure at high concentration.31,32 Therefore, the study on the MCPP at the single molecular level in water was necessary for exploring its potential biological applications. Single-molecule force spectroscopy (SMFS) has been considered as a powerful tool for investigating the mechanical chemistry of single polymer chains (e.g., force-induced conformational change in Fig. 4a).33,34 The force–extension (F–E) curves in Fig. 4b show the mechanical properties of four polymers in water. It is worth noting that longer PEG chains can be easily handled by the tip of atomic force microscopy, thus commercial PEG with a molecular weight of 35 kDa was used as the reference in this study. Compared with other three polymers, a larger force was required to stretch PEG from the normalized extension of 0.6 to 0.9. The reason for this can be attributed to the conformational change in PEG35k chains as reported in literature studies31,32 (Fig. 4a, top). During the stretching processes, PEG35k was transformed from a water-bridged helix to an all-trans conformation, and the bridges were broken simultaneously with an energy consumption of about 7.1 kJ mol−1 (Fig. 4c). However, this structural transition was not observed in other two typical MCPPs (MCPP4k (I−) and MCPP (I−)) (Fig. 4a, bottom, and Fig. S5†), where both these typical polymers showed the same F–E curves during the whole stretching process (Fig. 4b). Thus, this indicated that one or more rigid picolinium units could disturb the formation of helix structures in a flexible PEG chain and it was in accordance with the measurement of Tg and Td as shown in Fig. S3.† Caging picolinium units into MCPP chains made them more rigid than PEG35k, generating an extended conformation. On the other hand, the effect of a specific ion on the mechanical properties was studied by ion-exchanging I− to OTf− in MCPP chains. Fig. 4c shows that, compared with MCPP (I−), there was less force required to stretch the MCPP (OTf−) in the same normalized extension range, with an energy difference of about 4.5 kJ mol−1 (Fig. 4b and c). This difference can be assigned for the hydrophobicity of OTf−. As a consequence, there was a less tightly bound hydration shell formed around the hydrophobic MCPP (OTf−), and the weak hydration made the chains loose in water.
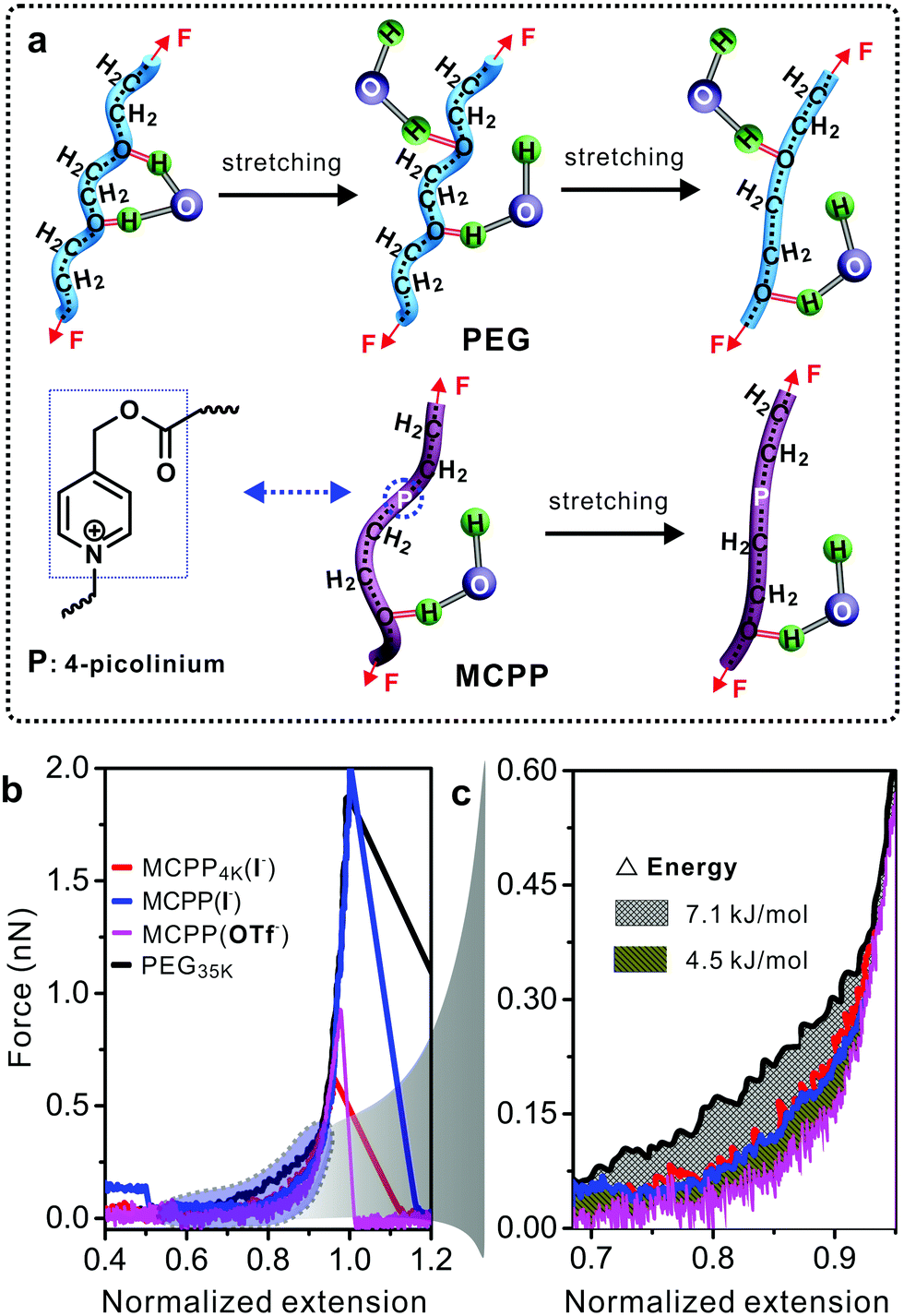 | ||
| Fig. 4 (a) Schematic of the hierarchy of elastic responses in single pure PEG and MCPP chains upon force stretching in water. (b) The comparison of normalized F–E curves of four polymers in water. (c) The partial F–E curves in (b). | ||
As a typical “stealth” polymer, PEG can increase the plasma residence and therapeutic index of drugs, resulting in reduced protein immunogenicity, prolonged period of circulation in the blood, decreased drug toxicity and clearance, etc.35–37 As for the current study, we would like to know whether the MCPP can be employed as a similar “stealth” polymer in a clinical or biological area. Notably, all of these processes mentioned above depend critically on the ability of the “stealth” polymer to pass through cell membranes. Therefore, it is important to investigate how the MCPP and degradation products pass through the membranes. As shown in Fig. 5a, the nanopore-contained lipid membranes and an aqueous solution of 3.0 M KCl in 10 mM Tris (pH = 7.2) were used as biomimetic cell membranes and the physiological environment, respectively. A bias voltage was applied to guide the MCPP and the photodegraded products through a-hemolysin (α-HL) nanopores.38,39
 | ||
| Fig. 5 (a) Schematic of the nanopore-based single-molecule analysis. (b) Representative single-channel current traces of MCPP and degradation products passing through the α-HL pores in an aqueous solution of 3 M KCl and10 mM Tris (pH = 7.2) at 60 mV. A histogram of bound currents and dwell times for (c and d) MCPP and (e and f) degradation products. | ||
By analyzing the current traces of samples as shown in Fig. 5b, it was found that for the MCPP, the events of passing through the pores can be classified into three levels, the count ratio was 32.3%:25.9%:41.8% and the dwell times were larger than 0.1 ms (Fig. 5c and d). It meant that there were ca. three types of MCPPs with different lengths in the sample. Furthermore, when increasing the bias voltage, the count ratio of three levels was changed to 20.4%:20%:59.6%, and the longest dwell time decreased from 0.74 ms to 0.538 ms (Fig. S6a–c†). The reason for this can be found in the positive-charged picolinium caging into the neutral chains, resulting in a bias voltage-sensitive MCPP. Consequently, increasing voltage (or current) can accelerate the MCPP to pass through the pores. This result was further confirmed by the control experiment of commercial non-charged PEG2k (Fig. 5e and f, and Fig. S7†): PEG2k was neutral so by varying the bias voltages applied, the changes were not clearly observed both in the events and count ratios. After the photodegradation of the MCPP, the events were significantly decreased, which was very similar to the blank experiment (without the polymer as shown in Fig. S6d†). Simultaneously, the count ratio was changed to 68.5%:24.2%:7.3%, and the dwell time for most of the samples passing through the pores was only 0.01 ms that was much shorter than that of the as-prepared MCPP (0.74 ms). This result indicated that the MCPP was photodegraded into products with low molecular weight. The above results indicated that both the MCPP and its degradation product could pass through the cell membranes by applying a bias voltage in the physiological environment.
More importantly, the cytotoxicities of MCPP and degradation products were tested at various concentrations using bone mesenchymal stem cells (BMSCs) that were collected from the redundant cancellous bone amputated during orthognathic surgery at Peking University Hospital of Stomatology in China. The quantification of the cytotoxic response was performed using the MTT assay. As shown in Fig. 6a, the cell viabilities of human BMSCs were all over 100% after 24 h of co-incubation with various amounts of MCPP, even when the concentration was increased to 500 μg mL−1. With the increase in time to 48 h and 72 h, the cell viabilities of most samples were still over 100%, except the sample with the extremely high content of MCPP (500 μg mL−1 at 48 h and over 50 μg mL−1 at 72 h). Moreover, the cell viabilities were all over 80% even after 72 h of co-incubation with the extremely high content of MCPP (500 μg mL−1). These results indicate that our MCPP not only has very low cytotoxicity but is also able to promote the cell proliferation at low concentration. For the degradation products, the cell proliferation was not observed anymore, and the cell viability was maintained ca. 100% at low concentrations (5 and 50 μg mL−1). When further increasing the concentration, the cell viability was decreased to >90% at 100 μg mL−1 (Fig. 6b). These results indicated that MCPP and degradation products possessed low cytotoxicity, showing potential applications in biological areas.
 | ||
| Fig. 6 Relative cell viability of BMSCs after the treatment with different concentrations of (a) MCPP and (b) the degradation products (n = 3). (c) The number of cells on bare (black open stars), MCPP modified (red spheres) and photodegraded (blue solid squares) substrates with different incubation times (n = 3). Optical images of BMSCs cells on (d and e) MCPP modified and (f) photodegraded substrates with predetermined incubation times. | ||
As a proof-of-concept, taking the advantage of the photodegradability and low toxicity of MCPP, a photo-switchable surface fabricated by surface-grafting of MCPP was used to demonstrate the potential applications of MCPP in cell proliferation, where the MCPP brushes were covalently grafted onto the substrates via the combination of thiol–ene click reaction and interfacial condensed polymerization (Fig. S8†). The as-prepared samples were tested for cell proliferation by using BMSCs which were contact-dependent for survival. The bare substrates were chosen as the negative control. To compare the performance of cell proliferation among these two samples, the same amounts of BMSCs were first seeded on each sample surface. As the incubation proceeded, the number of cells on both bare and MCPP modified surfaces was increased as shown in Fig. 6c–e and Fig. S9.† However, after 72 h of incubation, the ratio of the cell number of these two samples (MCPP modified surface:bare surface) increased dramatically and reached about 2:1 (Fig. 6c), indicating that the ability of MCPP modified samples to enhance cell proliferation outperformed the bare samples. Interestingly, the cell numbers on the MCPP modified samples after light irradiation increased slowly, with cell numbers similar to those on the bare substrates (Fig. 6c and f). The reason for this was attributed for the positive picolinium-contained MCPP. Such a photolabile polycation can significantly promote the cell attachment and growth on substrates.40–42 All these results indicated the good light-responsiveness of the MCPP layer, whose cell proliferation could be easily erased by light irradiation. These photo-switchable properties with selectable capability for enhancing cell proliferation may find applications in different areas of cell biology.
Conclusions
We have reported a main-chain visible-light-labile polymer by caging the photocleavable picolinium units into its backbones. The MCPP is degraded into small molecules with the catalysis of Ru(II)/ASC upon the blue light (452 nm) irradiation. Such a novel photodegradable MCPP shows several unique advantages as follows: (i) the synthesis of the MCPP is very simple offering great potential for medical applications. (ii) The MCPP and its degradation products can pass through the biomimetic cell membranes in a controlled fashion, thus showing potential applications in drug delivery. (iii) Nontoxic MCPP brushes can be easily grafted onto arbitrary substrates and the surface properties are photo-switchable, which are useful for enhancing cell proliferation selectively. Taking full advantage of these unique properties, it is anticipated that such an efficient main-chain visible-light-labile picolinium-caged polymer is a potential candidate with applications in the fields of materials, chemistry and biology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the NSFC (21604069, 21604013), the Natural Science Foundation of Shaanxi Province (2016JQ5035), and the Northwest University Graduate Innovation and Creativity Funds (YZZ17108) for the financial support of this work.Notes and references
- Y. Zhao, Macromolecules, 2012, 45, 3647–3657 CrossRef CAS.
- Q. Yan, D. Han and Y. Zhao, Polym. Chem., 2013, 4, 5026–5037 RSC.
- H. Zhao, E. S. Sterner, E. B. Coughlin and P. Theato, Macromolecules, 2012, 45, 1723–1736 CrossRef CAS.
- G. Pasparakis, T. Manouras, P. Argitis and M. Vamvakaki, Macromol. Rapid Commun., 2012, 33, 183–198 CrossRef CAS PubMed.
- T. E. Brown, I. A. Marozas and K. S. Anseth, Adv. Mater., 2017, 29, 1605001 CrossRef PubMed.
- S. Chatani, C. J. Kloxin and C. N. Bowman, Polym. Chem., 2014, 5, 2187–2201 RSC.
- C. A. Boyer and G. M. Miyake, Macromol. Rapid Commun., 2017, 38, 1700327 CrossRef PubMed.
- H.-C. Wang, Y. Zhang, C. M. Possanza, S. C. Zimmerman, J. Cheng, J. S. Moore, K. Harris and J. S. Katz, ACS Appl. Mater. Interfaces, 2015, 7, 6369–6382 CAS.
- V. X. Truong, F. Li, F. Ercole and J. S. Forsythe, Chem. Commun., 2017, 53, 12076 RSC.
- C. K. Arakawa, B. A. Badeau, Y. Zheng and C. A. DeForest, Adv. Mater., 2017, 29, 1703156 CrossRef PubMed.
- B. Yan, J.-C. Boyer, D. Habault, N. R. Branda and Y. Zhao, J. Am. Chem. Soc., 2012, 134, 16558–16561 CrossRef CAS PubMed.
- Z. C. Smith, D. M. Meyer, M. G. Simon, C. Staii, D. Shukla and S. W. Thomas, Macromolecules, 2015, 48, 959–966 CrossRef CAS.
- K. J. Arrington, J. B. Waugh, S. C. Radzinski and J. B. Matson, Macromolecules, 2017, 50, 4180–4187 CrossRef CAS.
- J. Dong, Z. Xun, Y. Zeng, T. Yu, Y. Han, J. Chen, Y.-Y. Li, G. Yang and Y. Li, Chem. – Eur. J., 2013, 19, 7931–7936 CrossRef CAS PubMed.
- W. Fan, X. Tong, Q. Yan, S. Fu and Y. Zhao, Chem. Commun., 2014, 50, 13492–13494 RSC.
- G. Liu, W. Liu and C.-M. Dong, Polym. Chem., 2013, 4, 3431–3443 RSC.
- F. Fan, L. Wang, F. Li, Y. Fu and H. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 17004–17010 CAS.
- L. Li, X.-X. Deng, Z.-L. Li, F.-S. Du and Z.-C. Li, Macromolecules, 2014, 47, 4660–4667 CrossRef CAS.
- L. García-Fernández, C. Herbivo, V. S. M. Arranz, D. Warther, L. Donato, A. Specht and A. d. Campo, Adv. Mater., 2014, 26, 5012–5017 CrossRef PubMed.
- J. B. Borak and D. E. Falvey, J. Org. Chem., 2009, 74, 3894–3899 CrossRef CAS PubMed.
- Z. Liu, Q. Lin, Q. Huang, H. Liu, C. Bao, W. Zhang, X. Zhong and L. Zhu, Chem. Commun., 2011, 47, 1482–1484 RSC.
- C. Sundararajan and D. E. Falvey, J. Org. Chem., 2004, 69, 5547–5554 CrossRef CAS PubMed.
- J. Kabatc and J. Pączkowski, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 576–588 CrossRef CAS.
- Y. Yu, X. Kang, X. Yang, L. Yuan, W. Feng and S. Cui, Chem. Commun., 2013, 49, 3431–3433 RSC.
- T. Zhou, Y. Lei, H. Zhang, P. Zhang, C. Yan, Z. Zheng, Y. Chen and Y. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 23431–23436 CAS.
- J. B. Borak, H.-Y. Lee, S. R. Raghavan and D. E. Falvey, Chem. Commun., 2010, 46, 8983–8985 RSC.
- J. L. Cape, J. B. Edson, L. P. Spencer, M. S. DeClue, H.-J. Ziock, S. Maurer, S. Rasmussen, P.-A. Monnard and J. M. Boncella, Bioconjugate Chem., 2012, 23, 2014–2019 CrossRef CAS PubMed.
- C. Sundararajan and D. E. Falvey, J. Am. Chem. Soc., 2005, 127, 8000–8001 CrossRef CAS PubMed.
- A. Eftekhari and T. Saito, Eur. Polym. J., 2017, 90, 245–272 CrossRef CAS.
- J. Wu, C. Zhao, W. Lin, R. Hu, Q. Wang, H. Chen, L. Li, S. Chen and J. Zheng, J. Mater. Chem. B, 2014, 2, 2983–2992 RSC.
- F. Oesterhelt, M. Rief and H. E. Gaub, New J. Phys., 1999, 1, 6 CrossRef.
- B. Heymann and H. Grubmüller, Chem. Phys. Lett., 1999, 307, 425–432 CrossRef CAS.
- S. Cui, J. Yu, F. Kühner, K. Schulten and H. E. Gaub, J. Am. Chem. Soc., 2007, 129, 14710–14716 CrossRef CAS PubMed.
- Z. Luo, A. Zhang, Y. Chen, Z. Shen and S. Cui, Macromolecules, 2016, 49, 3559–3565 CrossRef CAS.
- D. Pfister and M. Morbidelli, J. Controlled Release, 2014, 180, 134–149 CrossRef CAS PubMed.
- N. K. Singh and D. S. Lee, J. Controlled Release, 2014, 193, 214–227 CrossRef CAS PubMed.
- A. Kolate, D. Baradia, S. Patil, I. Vhora, G. Kore and A. Misra, J. Controlled Release, 2014, 192, 67–81 CrossRef CAS PubMed.
- Y. Wang, F. Yao and X.-f. Kang, Anal. Chem., 2015, 87, 9991–9997 CrossRef CAS PubMed.
- F. Yao, J. Duan, Y. Wang, Y. Zhang, Y. Guo, H. Guo and X. Kang, Anal. Chem., 2015, 87, 338–342 CrossRef CAS PubMed.
- R. Liu, X. Chen, S. H. Gellman and K. S. Masters, J. Am. Chem. Soc., 2013, 135, 16296–16299 CrossRef CAS PubMed.
- X. Hao, Q. Li, J. Lv, L. Yu, X. Ren, L. Zhang, Y. Feng and W. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 12128–12140 CAS.
- R. Liu, K. S. Masters and S. H. Gellman, Biomacromolecules, 2012, 13, 1100–1105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, supporting figures and NMR spectra. See DOI: 10.1039/c7py01844d |
| This journal is © The Royal Society of Chemistry 2018 |