
Chitosan hydrogelation with a phenothiazine based aldehyde: a synthetic approach toward highly luminescent biomaterials†

Abstract
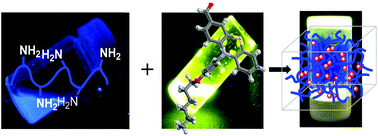
Pure organic luminescent hydrogels were synthesised using the condensation reaction of amino groups of chitosan with a photoactive aldehyde bearing a phenothiazine moiety. The hydrogels were structurally and supramolecularly characterized by FTIR and 1H-NMR spectroscopy, X-ray diffraction and polarized light microscopy. It was concluded that hydrogelation occurred due to the self-ordering of the chitosan segments grafted with pendant phenothiazine imines into ordered clusters, which play the role of crosslinking nodes. Scanning electron microscopy revealed microporous morphology with very thin pore walls. The ordered clusters immobilized in the polycationic chitosan network were responsible for high quantum efficiency, reaching the value of 51% in the xerogel state. The hydrogels formed flexible, transparent films with high mechanical toughness. Besides the remarkable optical and mechanical properties, the importance of the hydrogels is increased by their eco-design, based on chitosan from renewable resources and bio-friendly phenothiazine.
- This article is part of the themed collection: Frontiers in Supramolecular and Macromolecular Science symposia
 Please wait while we load your content...
Please wait while we load your content...

