DOI:
10.1039/C7PY01516J
(Paper)
Polym. Chem., 2018,
9, 98-107
Aluminium-catalyzed terpolymerization of furfuryl glycidyl ether with epichlorohydrin and ethylene oxide: synthesis of thermoreversible polyepichlorohydrin elastomers with furan/maleimide covalent crosslinks†
Received
5th September 2017
, Accepted 14th November 2017
First published on 29th November 2017
Abstract
By using an alkylaluminium catalyst system i-Bu3Al/H3PO4/1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), the random copolymerization and terpolymerization of furfuryl glycidyl ether (FGE) with epichlorohydrin (ECH) and ethylene oxide (EO) have been achieved for the first time to afford a new family of furan functionalized polyepichlorohydrin elastomers. The composition of the obtained FGE/ECH copolymers, FGE/EO copolymers and FGE/ECH/EO terpolymers coincided with the co-monomer feed ratio, and furan groups could be quantitatively and randomly incorporated into polyepichlorohydrin elastomers. Based on the Diels–Alder (DA) reaction of furan functionalized polyepichlorohydrin elastomers and 4,4′-methylenebis(N-phenylmaleimide), a new series of thermoreversible cross-linked polyepichlorohydrin elastomers was successfully synthesized. These thermoreversible DA cross-linked polyepichlorohydrin elastomers possess mechanical properties that can be tuned by adjusting the content of FGE units in the FGE/ECH/EO terpolymers. The mechanical properties of DA cross-linked polyepichlorohydrin elastomers were significantly better than those of traditional vulcanized polyepichlorohydrin elastomers, and they could be used as a structural material without reinforcing agents. More importantly, the DA cross-linked polyepichlorohydrin elastomers could be remolded and self-healed without loss of performance.
Introduction
Polyepichlorohydrin elastomers such as epichlorohydrin (ECH) homopolymers and ECH–ethylene oxide (EO) copolymers are typical functional polymeric materials with specific characteristics of interest for a wide range of applications in automotive, aerospace, printing and copying etc.1 However, these polyepichlorohydrin elastomers with excellent properties cannot be reprocessed and repaired after damage because of their irreversible cross-linking structure prepared by traditional vulcanization technologies. The introduction of reversible dynamic cross-linking structures to polyepichlorohydrin elastomers is highly desirable to construct a new type of reversible covalent crosslinking thermoreversible elastomers which possess excellent mechanical properties of cross-linked rubber under service conditions, while they would be remolded just like plastic materials after their product life. The Diels–Alder (DA) reaction between furan and maleimide is an excellent candidate for reversible polymer cross-linking because of relatively fast kinetics, mild reaction conditions, low coupling temperature and high decoupling temperature.2 So far, furan groups as crosslinking agents have already been randomly incorporated into epoxy resin,3,4 polyacrylates,5,6 polystyrene,7 polyketones,8 polybutadiene,9,10 ethylene/propylene rubber,11,12 ethylene/vinyl acetate rubber,13 and poly(propylene oxide).14 The incorporation of furan units as a block into poly(EO) by sequential living anionic ring opening polymerization has also been reported.15–18 However, the random copolymerization of FGE with ECH or terpolymerization of FGE with ECH and EO is still a difficult and challenging problem due to the reactivity ratios of monomers differing by several orders of magnitude.14,19–22
The alkylaluminium-based catalysts such as AlR3/H2O/third component23,24 and AlR3/H3PO4/Lewis base25,26 are well known to be efficient catalysts for the polymerization of epoxy monomers because of the ease of preparation, good stability, and versatility for various monomer substrates. In this paper, we have examined the potential of AlR3/H3PO4/Lewis base for the polymerization of FGE and its copolymerization with ECH and EO. By using i-Bu3Al/H3PO4/1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), the random copolymerization of FGE with ECH and EO and terpolymerization of FGE with ECH and EO have been achieved for the first time to give a new family of furan functionalized polyepichlorohydrin elastomers. The obtained furan functionalized polyepichlorohydrin elastomers were cross-linked in the presence of bismaleimide to afford a new type of thermoreversible polyepichlorohydrin elastomers with a dynamic thermoreversible cross-linked network; they demonstrated excellent mechanical properties, self-healing ability, and thermal recyclability.
Experimental section
Materials
Solvents were purified using an SPS-800 solvent purification system (MBraun) and stored over fresh Na chips. Epichlorohydrin (ECH) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were dried over CaH2 overnight, vacuum-transferred, and degassed by three freeze–pump–thaw cycles. Ethylene oxide (EO) was dried over CaH2 under 0 °C for 2 h, and vacuum-transferred into toluene at a concentration of 2.0 mol L−1. Furfuryl glycidyl ether (FGE) was synthesized according to the previously reported method,27 and purified by the same method with ECH. Pure phosphoric acid was diluted with ethyl ether to 1.25 mol L−1 under a pure nitrogen atmosphere. i-Bu3Al, 4,4′-methylenebis(N-phenylmaleimide) (BMI), Pb3O4, imidazolidine-2-thione (NA22), bis((dibutylcarbamothioyl)thio)nickel (NBC) and carbon black were used without further purification.
A typical procedure for the preparation of the i-Bu3Al/H3PO4/DBU catalyst (Table 1, run 2)
The i-Bu3Al/H3PO4/DBU catalyst was prepared in a dried ampoule bottle equipped with a magnetic stirrer under a pure nitrogen atmosphere. The ethyl ether solution of phosphoric acid (0.175 mmol, 1.25 mol L−1) was added dropwise to i-Bu3Al (0.500 mmol, 1.0 mol L−1 in toluene) at 0 °C under vigorous stirring. Then, the solution was warmed to room temperature and DBU (0.130 mmol) was added by a syringe. The solution was heated to 60 °C for 2 h under stirring and gave the colorless transparent i-Bu3Al/H3PO4/DBU catalyst solution.
Table 1 Copolymerization and terpolymerization of furfuryl glycidyl ether (FGE) with ethylene oxide (EO) and epichlorohydrin (ECH) by i-Bu3Al/H3PO4/DBUa
|
|
| Run |
H3PO4b |
DBUb |
ECHb |
EOb |
FGEb |
Yield (%) |
Compositionc (mol%) |
M
n
(×104) |
M
w/Mnd |
T
g
(°C) |
T
m
(°C) |
| ECH |
EO |
FGE |
|
Reaction conditions: i-Bu3Al, 0.5 mmol; monomer concentration, 2 mol L−1 in toluene; reaction time, 0.5 h; 25 °C; unless otherwise noted.
Molar ratio of i-Bu3Al.
Determined by 13C-NMR.
Determined by GPC in 1,2,4-trichlorobenzene at 135 °C against a polystyrene standard.
Determined by DSC.
Reaction time, 45 min
Not detected because of the very low solubility in THF, CHCl3, DMF, NMP and 1,2,4-trichlorobenzene.
|
| 1 |
0.2 |
0.26 |
20 |
20 |
0 |
Trace |
|
|
|
|
|
|
|
| 2 |
0.35 |
0.26 |
20 |
20 |
0 |
100 |
50 |
50 |
0 |
9.3 |
1.43 |
−39 |
— |
| 3 |
0.5 |
0.26 |
20 |
20 |
0 |
Trace |
|
|
|
|
|
|
|
| 4 |
0.35 |
0.10 |
20 |
20 |
0 |
43 |
20 |
80 |
0 |
0.9 |
1.91 |
−50 |
— |
| 5 |
0.35 |
0.40 |
20 |
20 |
0 |
100 |
50 |
50 |
0 |
24.1 |
1.62 |
−39 |
— |
| 6 |
0.35 |
0.50 |
20 |
20 |
0 |
100 |
50 |
50 |
0 |
33.3 |
1.72 |
−39 |
|
| 7 |
0.35 |
0.60 |
20 |
20 |
0 |
13 |
21 |
79 |
0 |
3.6 |
2.23 |
−49 |
— |
| 8 |
0.35 |
0.75 |
20 |
20 |
0 |
Trace |
|
|
|
|
|
|
|
| 9f |
0.35 |
0.26 |
0 |
0 |
40 |
100 |
0 |
0 |
100 |
n.d.g |
n.d. |
−27 |
— |
| 10 |
0.35 |
0.26 |
36 |
0 |
4 |
100 |
90 |
0 |
10 |
15.2 |
1.4 |
−23 |
— |
| 11 |
0.35 |
0.26 |
32 |
0 |
8 |
100 |
80 |
0 |
20 |
n.d. |
n.d. |
−24 |
— |
| 12 |
0.35 |
0.26 |
20 |
0 |
20 |
100 |
50 |
0 |
50 |
n.d. |
n.d. |
−25 |
— |
| 13 |
0.35 |
0.26 |
0 |
36 |
4 |
100 |
0 |
90 |
10 |
7.6 |
1.37 |
−49 |
54 |
| 14 |
0.35 |
0.26 |
0 |
32 |
8 |
100 |
0 |
80 |
20 |
n.d. |
n.d |
−43 |
38 |
| 15 |
0.35 |
0.26 |
0 |
20 |
20 |
100 |
0 |
50 |
50 |
n.d. |
n.d. |
−34 |
— |
| 16 |
0.35 |
0.26 |
4 |
4 |
32 |
100 |
10 |
10 |
80 |
n.d. |
n.d. |
−31 |
— |
| 17 |
0.35 |
0.26 |
10 |
10 |
20 |
96 |
25 |
25 |
50 |
n.d. |
n.d. |
−36 |
— |
| 18 |
0.35 |
0.26 |
14 |
14 |
12 |
100 |
35 |
35 |
30 |
n.d. |
n.d. |
−37 |
— |
| 19 |
0.35 |
0.26 |
16 |
16 |
8 |
100 |
40 |
40 |
20 |
12.5 |
1.51 |
−39 |
— |
| 20 |
0.35 |
0.26 |
18 |
18 |
4 |
100 |
45 |
45 |
10 |
12.7 |
1.56 |
−39 |
— |
| 21 |
0.35 |
0.26 |
19 |
19 |
2 |
100 |
47 |
47 |
6 |
10.5 |
1.48 |
−39 |
— |
| 22 |
0.35 |
0.26 |
38 |
38 |
4 |
100 |
47 |
48 |
5 |
25.7 |
1.44 |
−39 |
— |
| 23 |
0.35 |
0.26 |
2.8 |
11.2 |
26 |
100 |
7 |
28 |
65 |
n.d. |
n.d. |
−31 |
— |
| 24 |
0.35 |
0.26 |
4.8 |
19.2 |
16 |
100 |
12 |
48 |
40 |
n.d. |
n.d. |
−34 |
— |
| 25 |
0.35 |
0.26 |
12 |
24 |
4 |
100 |
30 |
60 |
10 |
10.4 |
1.52 |
−45 |
— |
| 26 |
0.35 |
0.26 |
24 |
12 |
4 |
100 |
60 |
30 |
10 |
12.2 |
1.32 |
−31 |
— |
A typical procedure for the copolymerization of ECH and EO (Table 1, run 2)
Polymerization was carried out in a dried ampoule bottle equipped with a magnetic stirrer under a pure nitrogen atmosphere. Toluene (5 mL), ECH (10 mmol), and EO (10 mmol, 2.0 mol L−1 in toluene) were injected into the ampoule bottle by a syringe; then the i-Bu3Al/H3PO4/DBU catalyst solution prepared as described above was injected into the ampoule bottle while stirring with a magnetic stirrer. After the polymerization was carried out at 25 °C for 0.5 h, a viscous toluene solution of the ECH/EO (ECO) copolymer was obtained. The solution was poured into a larger amount of n-hexane to precipitate the ECO copolymer, which was washed with methanol and dried under vacuum at 40 °C to a constant weight. The dried ECO copolymer was used for chemical analysis. The ECO copolymer solution for vulcanization crosslinking was scale-up synthesized in a 250 mL stainless autoclave by the same procedures.
A typical procedure for the terpolymerization of ECH, EO and FGE (Table 1, run 23)
Polymerization was carried out in a dried ampoule bottle equipped with a magnetic stirrer under a pure nitrogen atmosphere. Toluene (6 mL), ECH (8 mmol), EO (8 mmol, 2.0 mol L−1 in toluene) and FGE (4 mmol) were injected into the ampoule bottle by a syringe; then the i-Bu3Al/H3PO4/DBU catalyst solution prepared as described above was injected into the ampoule bottle while stirring with a magnetic stirrer. After the polymerization was carried out at 25 °C for 0.5 h, a viscous toluene solution of FGE/ECH/EO (ECO/FGE20) terpolymer was obtained. The solution was poured into a larger amount of n-hexane to precipitate the ECO/FGE20 terpolymer, which was washed with methanol and dried under vacuum at 40 °C to a constant weight. The dried ECO/FGE20 terpolymer was used for chemical analysis. The ECO/FGE20 terpolymer solution for the Diels–Alder (DA) cross-linking reaction was scale-up synthesized in a 250 mL stainless autoclave by the same procedures. Other ECO/FGEm terpolymers and terpolymer solutions for DA crosslinking were obtained by the same procedure, where m is defined as molar percentage content of FGE in the FGE/ECH/EO terpolymers, and the molar ratio of ECH and EO is 1.
Diels–Alder cross-linking reaction of FGE/ECH/EO terpolymers (ECO/FGEm) and preforming
The antioxidant NBC (2.0 wt% of the polymer) and BMI (0.5 equivalent of furan groups in terpolymers) were added into the toluene solutions of ECO/FGEm terpolymers prepared as described above. After homogeneous mixing, the resulting mixtures were poured into a larger amount of n-hexane to precipitate ECO/FGEm terpolymers mixed with BMI (ECO/FGEm/BMI0.5m), which were washed with methanol and dried under vacuum at 40 °C to a constant weight. The ECO/FGEm/BMI0.5m terpolymers were compression molded for 30 min at 155 °C and 100 bar. Then the resulting ECO/FGEm/BMI0.5m terpolymer plaques were crosslinked for 8 h at 70 °C in an oven to afford red28 DA cross-linked materials (DA_ECO/FGEm/BMI0.5m). The cross-linking degree of DA_ECO/FGEm/BMI0.5m could be tuned by adjusting the content of FGE and BMI.
Vulcanization of cross-linking of ECH/EO copolymers and preforming
The antioxidant NBC (2.0 wt% of the polymer), Pb3O4 (6.0 wt% of the polymer), the cross-linking agent NA22 (0.8 wt% of the polymer) were added into a toluene solution of the ECH/EO copolymer prepared as described above. After homogeneous mixing, the resulting mixture was poured into a larger amount of n-hexane to precipitate the ECH/EO copolymer mixed with NA22 (ECO/NA220.8), which was washed with methanol and dried under vacuum at 40 °C to a constant weight. The products were compression molded and vulcanized for 35 min at 155 °C and 100 bar to afford the vulcanization cross-linked material vulcanized_ECO/NA220.8. Other vulcanized_ECO/NA22n materials were obtained by using the same method, where n is defined as weight content of NA22 in the ECH/EO copolymers (the molar ratio of ECH and EO is 1). The cross-linking degree of vulcanized_ECO/NA22n could be tuned by adjusting the content of NA22. Carbon black reinforced material (vulcanized/carbon) was also obtained by the same method but with extra 40 wt% carbon black.
Kinetic experiments of ECH/EO, EO/FGE and ECH/FGE copolymerization
In an NMR sample tube, ECH (260 μmol) and FGE (260 μmol) were added to 1.0 mL of deuterated toluene. After homogeneous mixing, 0.1 mL of the catalyst solution (containing 13 μmol of i-Bu3Al; the mole ratio of i-Bu3Al/H3PO4/DBU is 1/0.6/0.26) was added. The NMR sample tube was then quickly placed in a Bruker AC-400 NMR spectrometer, and data were collected until the signals of monomers disappeared. The NMR spectrometer was held at 25 °C and collected scans at periodic time intervals. The kinetic data and time-dependent composition of ECH/FGE copolymerization were used to calculate reactivity ratio adopted a Nonterminal Model of Chain Copolymerization.29 The same procedure was used to obtain the reactivity ratio of ECH/EO and EO/FGE copolymerization.
Characterization
The 13C-NMR spectra of ECH/EO, FGE/ECH, FGE/EO copolymers and FGE/ECH/EO terpolymers were recorded on a Bruker AC-400 spectrometer in C2D2Cl4 at 25 °C. The molecular weights and molecular weight distributions of ECH/EO, FGE/ECH, FGE/EO copolymers and FGE/ECH/EO terpolymers were determined at 135 °C by high temperature gel permeation chromatography (HT-GPC) on an Agilent PL-GPC220 GPC apparatus. 1,2,4-Trichlorobenzene was employed as an eluent at a flow rate of 1.0 mL min−1. The calculation was made with a polystyrene standard. The DSC measurements of ECH/EO, FGE/ECH, FGE/EO copolymers, FGE/ECH/EO terpolymers and DA crosslinked materials were performed using a TA DSC Q20 instrument under a nitrogen atmosphere. Any thermal history difference of the polymers was eliminated by first heating to 200 °C (100 °C for DA crosslinked materials) at 10 °C min−1, cooling to −70 °C at 10 °C min−1, then heating to 200 °C (100 °C for DA crosslinked materials) at 10 °C min−1, and then recording the second scan results. Fourier transform infrared (FT-IR) spectra were acquired on a Nicolet 6700 Flex spectrometer. The film was prepared by dissolving and mixing ECO/FGE20 terpolymer with BMI (0.5 equivalent of furan groups in the polymer) in dichloromethane, and then a volatile solvent in KBr pellets. The film was heated to 70 °C to detect DA reaction and 150 °C to detect retro-DA reaction. The tensile properties of the DA and vulcanization cross-linked materials were measured using an Instron 5576A instrument at room temperature with a humidity of about 30% at a crosshead speed of 10 mm min−1. Dumbbell specimens with a length of 35 mm, a width of 6 mm and a thickness of 1 mm were cut from cross-linked plaque. Data analyses were based on five measurements on each sample performed under the same conditions. The cross-link density ([XLD]S in moles per gram) of crosslinked materials was determined from swelling experiments in THF. Approximately 50 mg (m1) of the dried, cross-linked polymer was weighed into a 30 mL bottle and immersed in 25 mL of THF until equilibrium swelling (1 day) was reached (m2). Subsequently, the swollen sample was dried in a vacuum oven at 40 °C until a constant weight was reached (m3). The weights of the swollen and dried samples were used to calculate the swelling ratio, gel fraction and apparent cross-link density using the Flory–Rehner equation:
| [XLD]S = [ln(1 − ϕ) + ϕ + χϕ2]/[−2ρECOVTHFϕ1/3]; |
| ϕ = (m3/ρECO)/[(m3/ρECO) + (m2 − m1)/ρTHF]; |
| Swelling ratio = (m2/m1 − 1); Gel fraction = m3/m1; |
where ρTHF is the density of THF, ρECO is the density of the polymer, VTHF is the molar volume of THF, and χ is the interaction parameter.
Theoretical cross-link density was calculated according to the following formula:
| [XLD]S = x/(xMFGE + yMECH + zMEO + 0.5xMBMI) |
where
x,
y, and
z are the molar contents of FGE, ECH, and EO units in the terpolymer calculated by NMR analysis, respectively;
MFGE,
MECH,
MEO, and
MBMI are the molar weights of FGE, ECH, EO and BMI.
Results and discussion
Copolymerization and terpolymerization of furfuryl glycidyl ether with epichlorohydrin and ethylene oxide
The catalytic system i-Bu3Al/H3PO4/Lewis base is a classical catalyst for the polymerization of ECH and other epoxides.25,26 The composites of this ternary catalyst have a serious impact on polymerization activity, copolymerization behaviour and stereo-selectivity.25,26 To seek a highly efficient catalyst for the random copolymerization of FGE with ECH and EO, the copolymerization of ECH with EO was first investigated by using different compositions of the i-Bu3Al/H3PO4/DBU catalyst. When the H3PO4/i-Bu3Al ratio was 0.35, the copolymerization was completed within 30 min, and the yield of the ECH/EO copolymer was as high as 100% (Table 1, run 2). When the H3PO4/i-Bu3Al ratio was higher or lower than 0.35, the catalyst almost lost its copolymerization activity (Table 1, runs 1 and 3). The molar ratio of DBU/i-Bu3Al had also a serious effect on copolymerization activity. When the range of the DBU/i-Bu3Al mole ratio was between 0.26 and 0.5, the copolymerization was completed within 30 min with the yield of ECH/EO copolymers as high as 100%, while the molecular weight of ECH/EO copolymers increased with DBU/i-Bu3Al ratio (Table 1, runs 2, 5 and 6). When the DBU/i-Bu3Al mole ratio was higher than 0.5 or lower than 0.26, the copolymerization activity dramatically decreased (Table 1, runs 4 and 7). When the DBU/i-Bu3Al ratio was higher than 0.75, the catalyst lost its catalytic activity (Table 1, run 8). These results suggested that the formation of a high active site required quantitative reaction between i-Bu3Al, H3PO4 and DBU. Firstly, the reaction of i-Bu3Al with H3PO4 at the ratio of 1/0.35 might afford dialkylaluminium phosphate [(i-Bu2AlO)3P(O)]n;30–32 then, the quantitative DBU coordinated to aluminium of dialkylaluminium phosphate could give a high active site. In our experiment range, when the molar ratio of DBU/i-Bu3Al was lower than 0.26, only part of the active center can be formed which resulted in lower activity; when the molar ratio of DBU/i-Bu3Al was higher than 0.26, the excessive DBU would be further coordinated, which could result in partial catalyst deactivation; when the molar ratio of DBU/i-Bu3Al was up to 0.75, the seriously excess DBU would destroy all the active center and resulted in no polymerization ability. The copolymerization activity reached the highest values at i-Bu3Al/H3PO4/DBU = 1/0.35/0.26, which suggested that the high active site might be a DBU coordinated with four aluminium atoms of dialkylaluminium phosphate.
Then the copolymerization of ECH and EO with different feed composition was investigated by using i-Bu3Al/H3PO4/DBU (1/0.35/0.26) to examine the ability of the catalyst to control the composition and degree of randomness of ECH/EO copolymers (Table S1, ESI†). The copolymerization of ECH and EO was completed within 30 min, irrespective of the monomer feed ratio. The compositions of ECH/EO copolymers coincided with the co-monomer feed ratio, suggesting that both monomers were completely incorporated into ECH/EO copolymers. The monomer reactivity ratios rECH = 0.74 and rEO = 1.41 suggested that the formation of ECH/EO random copolymers is preferred in the present ECH/EO copolymerization (Fig. S9A, ESI†). The microstructures of ECH/EO copolymers were characterized by 13C-NMR analyses (Fig. S1, ESI†); the results also showed that ECH/EO copolymers had a random monomer sequence distribution. All of these ECH/EO copolymers with different compositions showed a single glass transition temperature (Tg) which varied with the ECH/EO ratio. The Tgs of ECH/EO copolymers with ECH content = 20–80 mol% fell in the range of −46 °C to −28 °C, which are between homopoly(ECH) (−22 °C) and homopoly(EO) (−53 °C), and increased linearly with increasing ECH content, in agreement with the random distribution of ECH and EO in the copolymers. These results of ECH and EO copolymerization showed that the catalyst system i-Bu3Al/H3PO4/DBU (1/0.35/0.26) served as an excellent catalyst for the random copolymerization of ECH and EO.
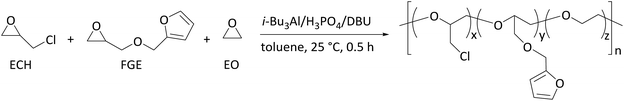
Based on the high activity and random copolymerization behavior of i-Bu3Al/H3PO4/DBU in the polymerization of ECH and EO, we then investigated the incorporation of furfuryl glycidyl ether (FGE) into ECH homopolymers, EO homopolymers, and ECH/EO random copolymers. Some representative results are summarized in Table 1. At first, the homopolymerization of FGE by using i-Bu3Al/H3PO4/DBU afforded 3.0 g (100% yield) homopoly(FGE) in 45 min (Table 1, run 9), which suggested that furan groups had no effect on the catalyst activity. Similarly to the copolymerization of ECH and EO, the two-component copolymerization of FGE and ECH (Table 1, runs 10–12) and that of FGE and EO (Table 1, runs 13–15) have been achieved to afford the corresponding random copolymers with high activity and controllable composition (rEO/rFGE = 2.42/0.21, rECH/rFGE = 1.34/0.71, Fig. S9B and C, ESI†). The 13C-NMR spectra of FGE/ECH copolymers with a FGE content of 50 mol% and 10 mol% are shown in Fig. 1A and B, respectively. The similar intensities of peaks of FGE–ECH sequences, FGE–FGE sequences, and ECH–ECH sequences were observed in the 13C-NMR spectrum of the FGE/ECH copolymer with a FGE content of 50 mol% (Fig. 1A). The isolated FGE units and ECH–ECH blocks were observed in the 13C-NMR spectrum of the FGE/ECH copolymer with a FGE content of 10 mol% (Fig. 1B). These results clearly suggested the random microstructure of FGE/ECH copolymers prepared by i-Bu3Al/H3PO4/DBU. The 13C-NMR spectra of FGE/EO copolymers with a FGE content of 50 mol% and 10 mol% are also shown in Fig. 1C and D. Similarly, the FGE/EO copolymer with a FGE content of 50 mol% contained FGE–EO sequences, FGE–FGE sequences, and EO–EO sequences. The FGE/EO copolymer with a FGE content of 10 mol% contained FGE–EO sequences and EO–EO sequences. The FGE/EO copolymers prepared by i-Bu3Al/H3PO4/DBU were also random microstructures. All of these novel FGE/ECH and FGE/EO copolymers with different compositions showed a single Tg, in agreement with the random distribution of two monomers in the copolymers.
 |
| | Fig. 1
13C-NMR spectra (100 MHz, 1,1,2,2-tetrachloroethane-d2, 25 °C) of FGE/ECH copolymers (A and B), FGE/EO copolymers (C and D), and a FGE/ECH/EO terpolymer (E). | |
In the presence of FGE, ECH and EO, the three-component copolymerization also took place rapidly irrespective of the monomer feed ratio within 30 min, also giving exclusively the corresponding FGE/ECH/EO terpolymers with 100% yield, high molecular weights (Mn = 10.5–25.7 × 104) and narrow molecular weight distributions (Mw/Mn = 1.3–1.5). The composition of FGE/ECH/EO terpolymers coincided with the co-monomer feed ratio, suggesting that furan groups were quantitatively incorporated into the terpolymers (Table 1, runs 16–26). An increase of the feeding amount of the monomers relative to the catalyst led to a significant increase of the molecular weights of the terpolymer products (Table 1, run 22). The 13C-NMR spectrum of a FGE/ECH/EO terpolymer with FGE content = 10 mol%, ECH content = 60 mol%, and EO content = 30 mol% is shown in Fig. 1E. It was found that these terpolymer products containing FGE–EO sequences, FGE–ECH sequences, ECH–EO sequences, ECH–ECH sequences, and EO–EO sequences are random terpolymers (Fig. S5, ESI†). All of these obtained FGE/ECH/EO terpolymers with FGE content = 5–80 mol%, ECH content = 7–60 mol% and EO content = 7–60 mol% showed a single Tg at −31 °C to −45 °C (Table 1, runs 16–26), which suggested the random distribution of FGE, ECH and EO units in the terpolymers. These polymerization results showed that i-Bu3Al/H3PO4/DBU served as an excellent catalyst for the random copolymerization and terpolymerization of FGE with ECH and EO to afford a new family of polyepichlorohydrin elastomers with a controllable number of pendant furan groups.
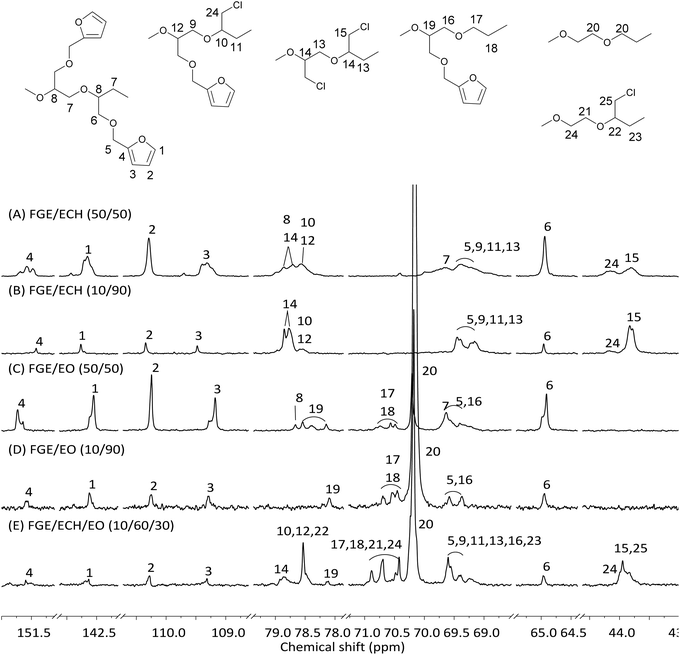
Cross-linking and retrocross-linking of furan functionalized polyepichlorohydrin elastomers
Based on the successful synthesis of polyepichlorohydrin elastomers with a controllable number of pendant furan groups, the DA cross-linking reaction of furan functionalized polyepichlorohydrin elastomers was investigated by directly mixing 4,4′-methylenebis(N-phenylmaleimide) (BMI) with a FGE/ECH/EO terpolymer solution after the end of polymerization, as shown in Scheme 1. FGE/ECH/EO terpolymers mixed with BMI (ECO/FGEm/BMI0.5m) precipitated from the polymerization solution were heated to 70 °C for 8 hours; a red32 DA cross-linked polyepichlorohydrin elastomer (DA_ECO/FGEm/BMI0.5m) was successfully obtained. The cross-linking structure of DA_ECO/FGEm/BMI0.5m almost decoupled when DA_ECO/FGEm/BMI0.5m was heated to 150 °C for 2 h (Fig. S11, ESI†).
 |
| | Scheme 1 Synthesis of DA cross-linked polyepichlorohydrin elastomers. | |
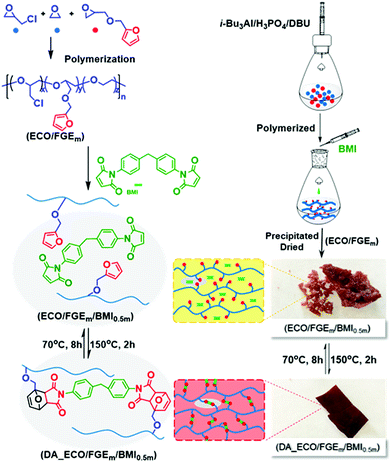
The gel fraction and cross-linking density of DA_ECO/FGEm/BMI0.5m materials are shown in Fig. 2. When the FGE content in FGE/ECH/EO terpolymers increased from 1 mol% to 20 mol%, the gel fraction of DA cross-linked material was above 92%, and the cross-linking density increased from 1.3 × 10−4 mol g−1 to 15.3 × 10−4 mol g−1. The cross-linking density of polyepichlorohydrin elastomers could be easily tuned by controlling the content of FGE in FGE/ECH/EO terpolymers. In addition, the cross-link density determined by swelling experiments was of the same order of magnitude as the theoretical value calculated by NMR analysis. These results in combination with the distribution of FGE units in FGE/ECH/EO terpolymers suggested that there was a random and uniform distribution of the intermolecular crosslinks throughout polyepichlorohydrin elastomers.
 |
| | Fig. 2 The gel fraction and cross-linking density of DA_ECO/FGEm/BMI0.5m materials with different FGE contents. | |
Thermal and mechanical properties of DA cross-linked polyepichlorohydrin elastomers
With the successful synthesis of DA cross-linked polyepichlorohydrin elastomers with different cross-linking densities, their thermal and mechanical properties were also determined by DSC, TGA and tensile tests. The decomposition temperatures of DA_ECO/FGEm/BMI0.5m materials defined by the onset point of 5% degradation were all over 280 °C, which demonstrated that these DA cross-linked polyepichlorohydrin elastomers possessed good thermostability (Fig. S23, ESI†). DSC curves of DA_ECO/FGEm/BMI0.5m materials are shown in Fig. 3. When the FGE content in FGE/ECH/EO terpolymers was lower than 10 mol%, DA_ECO/FGEm/BMI0.5m materials possessed a low Tg, which suggested that these materials showed good low-temperature resistance. When the FGE content in FGE/ECH/EO terpolymers was higher than 20%, the Tg of DA_ECO/FGEm/BMI0.5m materials disappeared because of high cross-linking degree.
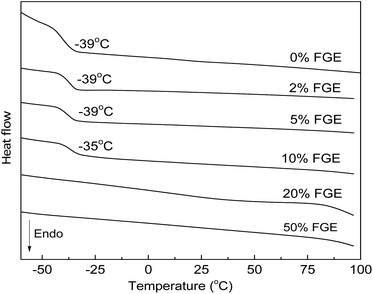 |
| | Fig. 3 DSC curves of DA_ECO/FGEm/BMI0.5m materials with different FGE contents. | |
The tensile stress–strain curves of DA_ECO/FGEm/BMI0.5m materials are shown in Fig. 4. First, the non-cross-linked FGE/ECH/EO terpolymers (Table S4, runs 2, ESI†) and the ECH/EO copolymers were similar in tensile strength (about 1.0 MPa), elongation at break (about 600%), and Young's modulus (about 0.1 MPa), which suggested that these non-cross-linked polymers were soft and weak rubbers. In sharp contrast, the cross-linked DA_ECO/FGE1/BMI0.5 material containing 1 mol% FGE possessed a tensile strength of 7.3 MPa, an elongation at break of 1221%, and a Young's modulus of 0.6 MPa, and the DA_ECO/FGE1/BMI0.5 materials has become a strong and tough material. Increasing FGE content to 3 mol% could increase the tensile strength of DA cross-linked polyepichlorohydrin elastomers up to 15.0 MPa, while the elongation at break still decreased to 504%. As we all know, traditional vulcanized ECH/EO copolymers with carbon black as a reinforcing agent (vulcanized/carbon) has a tensile strength 12.9 MPa, an elongation at break of 620%, and a Young's modulus of 2.0 MPa. The mechanical properties of the DA_ECO/FGE3/BMI1.5 material has exceeded traditional vulcanized rubbers with carbon black as a reinforcing agent, so the DA_ECO/FGE3/BMI1.5 material without reinforcing agents can be used as a structural material. On further increasing FGE content from 3 mol% to 10 mol%, the Young's modulus of DA cross-linked polyepichlorohydrin elastomers was increased from 2.1 MPa to 25.0 MPa (Table S4, run 9, ESI†), while the elongation at break was decreased from 504% to 58% with the tensile strength shifting around 12.0 MPa. Based on the above tensile testing, the DA cross-linked polyepichlorohydrin elastomers showed mechanical properties that can be tuned by changing FGE content with great freedom.
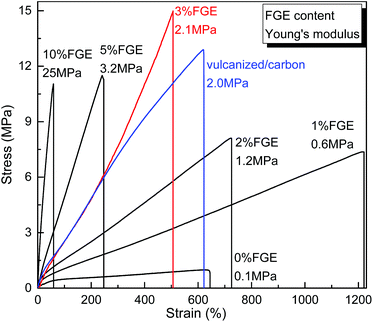 |
| | Fig. 4 Stress–strain curves and Young's modulus of DA_ECO/FGEm/BMI0.5m materials with different FGE contents. | |
The relationships of mechanical properties versus crosslinking density for DA cross-linked polyepichlorohydrin elastomers (DA_ECO/FGEm/BMI0.5m) and traditional vulcanized ECH/EO copolymer materials (ECO/NA22n) are also shown in Fig. 5. When the cross-linking density was 1.2 × 10−4 mol g−1, the tensile strength of traditional vulcanized ECH/EO copolymers reached the highest value of 7.3 MPa, and then decreased extremely with the increase of cross-linking density. Compared with the two methods of cross-linking, DA cross-linked polyepichlorohydrin elastomers possessed tensile strengths significantly higher than those of traditional vulcanized materials (Fig. 5A); not only that, DA cross-linked polyepichlorohydrin elastomers had similar excellent elongation at break and Young's modulus to traditional vulcanized materials (Fig. 5B and C). The uniform FGE distribution in FGE/ECH/EO terpolymers and uniform intermolecular DA cross-linking should be important factors for the excellent mechanical properties.
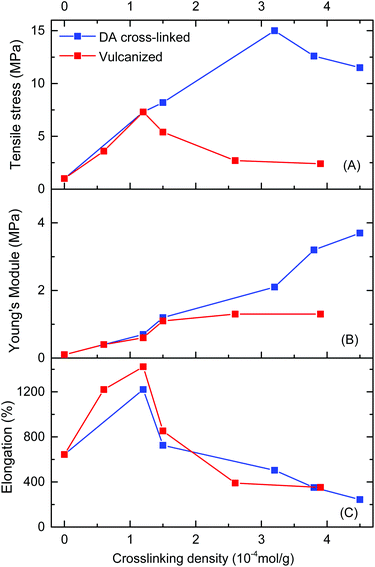 |
| | Fig. 5 Tensile strength (A), Young's modulus (B), and elongation at break (C) of DA_ECO/FGEm/BMI0.5m materials (blue) and vulcanized ECO/NA22n materials (red) with a series of cross-linking densities. | |
Thermal recyclability and self-healing ability of DA cross-linked polyepichlorohydrin elastomers
The DA cross-linked polyepichlorohydrin elastomers not only possessed excellent mechanical properties compared to traditional vulcanized rubber, but also possessed thermal recyclability because of reversible DA cross-linking structures. As shown in Fig. 6, the pieces of broken representative DA_ECO/FGE3/BMI1.5 material were reprocessed under a pressure of 100 bar for 2 h at 155 °C, and a solid film was re-formed again. Then the remolded film was cut into pieces, and the recycling process was repeated two times. The remolded film exhibited almost the same mechanical properties even through three generations of recycling (the range is for tensile strength 13.1–15.0 MPa, elongation 455.8%–520.7%, and Young’ modulus 2.0–2.1 MPa). The DA cross-linked polyepichlorohydrin elastomers not only can be used as a structural material without reinforcing fillers, but also could be repossessed like plastic materials.
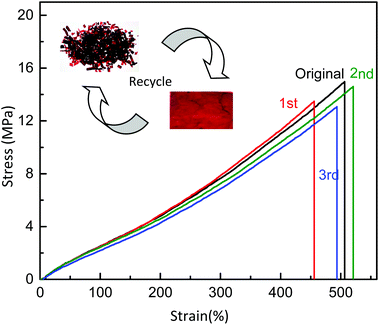 |
| | Fig. 6 Stress–strain curves of the thermal recyclability of the DA_ECO/FGE3/BMI1.5 material. | |
Except recyclability like plastic materials, the self-healing property of DA cross-linked polyepichlorohydrin elastomers based on retrocross-linking process was also examined. On the representative DA_ECO/FGE3/BMI1.5 material were created cracks of different widths by using a scalpel, and then the cracked film was heated at 150 °C for 3 h. Microscopic images indicated the cracks of about 100 μm width greatly decreased to 46 μm width (Fig. 7A and B), while smaller cracks such as 68 μm width cracks disappeared completely (Fig. 7C and D). This result showed that DA cross-linked polyepichlorohydrin elastomers can well repair themselves with an easy retrocross-linking process at high temperature. The self-healing process that is completed at room temperature would be more attractive than that at high temperature because it is friendly to heat-sensitive complex devices. The representative DA_ECO/FGE20/BMI1.5 material that has rich unreacted furan groups was chosen to achieve self-healing based on the further cross-linking of uncross-linked furan groups at room temperature.33 The cracks in the DA_ECO/FGE20/BMI1.5 material were filled with a chloroform solution of BMI; then the film was placed at room temperature for 3 days to finish the DA cross-linking reaction between uncross-linked furan groups in the DA_ECO/FGE20/BMI1.5 and the chloroform solution of BMI and then obtain the restored samples. The microscopic images of the scratched and restored film showed that the big cracks such as 130 μm width cracks could be completely healed at room temperature (Fig. 7E and F). Relying on the well-defined structures, the DA crosslinked polyepichlorohydrin elastomers possessed excellent self-healing ability not only at high temperature but also at room temperature.
 |
| | Fig. 7 Microscopic images for the self-healing process of DA cross-linked polyepichlorohydrin elastomers at high temperature and room temperature. DA_ECO/FGE3/BMI1.5 material with a scratch at room temperature (A and C), and self-healed film at 150 °C for 3 h (B and D); DA_ECO/FGE20/BMI1.5 material with a scratch at room temperature (E), and self-healed film at room temperature for 3 day (F). | |
Conclusions
We have demonstrated that the aluminium-based catalyst i-Bu3Al/H3PO4/DBU can serve as an excellent catalyst for the random copolymerization and terpolymerization of FGE with ECH and EO to afford a new family of furan functionalized polyepichlorohydrin elastomers. The composition of the obtained FGE/ECH copolymers, FGE/EO copolymers and FGE/ECH/EO terpolymers coincided with the co-monomer feed ratio, and furan groups could be quantitatively and randomly incorporated into polyepichlorohydrin elastomers. The thermoreversible cross-linked polyepichlorohydrin elastomers with furan/maleimide covalent crosslinks were successfully synthesized based on the DA reaction of furan functionalized polyepichlorohydrin and BMI. The obtained DA cross-linked polyepichlorohydrin elastomers possess mechanical properties that can be tuned by adjusting the content of FGE in the FGE/ECH/EO terpolymers. Compared with traditional vulcanized materials, DA cross-linked polyepichlorohydrin elastomers possessed higher tensile strength and similar excellent elongation at break and Young's modulus. The mechanical properties of the DA_ECO/FGE3/BMI1.5 material have exceeded traditional vulcanized materials with carbon black as a reinforcing agent. More importantly, the new DA cross-linked polyepichlorohydrin elastomers could be remolded and self-healed without loss of performance. This work has demonstrated that the well-defined DA cross-linked polyepichlorohydrin elastomers with excellent recycling properties and self-healing ability can be used as a structural material without reinforcing fillers, and provide a more energy-efficient, more environmentally friendly new method for the synthesis of cross-linked polyepichlorohydrin elastomers.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21674016, 21429201, 21034001, and U1508204).
Notes and references
-
R. Klingender, in Handbook of Specialty Elastomers, CRC Press, 1st edn, 2008, pp. 245–288 Search PubMed.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1–29 CrossRef CAS.
- A. M. Peterson, R. E. Jensen and G. R. Palmese, ACS Appl. Mater. Interfaces, 2010, 2, 1141–1149 CAS.
- P. A. Pratama, M. Sharifi, A. M. Peterson and G. R. Palmese, ACS Appl. Mater. Interfaces, 2013, 5, 12425–12431 CAS.
- W. Hu, Z. Ren, J. Li, E. Askounis, Z. Xie and Q. Pei, Adv. Funct. Mater., 2015, 25, 4827–4836 CrossRef CAS.
- E. Goiti, M. B. Huglin and J. M. Rego, Macromol. Rapid Commun., 2003, 24, 692–696 CrossRef CAS.
- M. P. Stevens and A. D. Jenkins, J. Polym. Sci., Part A: Polym. Chem., 1979, 17, 3675–3685 CrossRef CAS.
- Y. Zhang, A. A. Broekhuis and F. Picchioni, Macromolecules, 2009, 4, 1906–1912 CrossRef.
- E. Trovatti, T. M. Lacerda, A. J. Carvalho and A. Gandini, Adv. Mater., 2015, 27, 2242–2245 CrossRef CAS PubMed.
- J. Bai, H. Li, Z. Shi and J. Yin, Macromolecules, 2015, 48, 3539–3546 CrossRef CAS.
- L. M. Polgar, M. van Duin, A. A. Broekhuis and F. Picchioni, Macromolecules, 2015, 48, 7096–7105 CrossRef CAS.
- A. Wang, H. Niu, Z. He and Y. Li, Polym. Chem., 2017, 8, 4494–4502 RSC.
- L. M. Polgar, E. Hagting, W.-J. Koek, F. Picchioni and M. van Duin, Polymers, 2017, 9, 81–94 CrossRef.
- K. Roos, E. Dolci, S. Carlotti and S. Caillol, Polym. Chem., 2016, 7, 1612–1622 RSC.
- M. J. Barthel, T. Rudolph, S. Crotty, F. H. Schacher and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4958–4965 CrossRef CAS.
- C. Hörenz, T. Rudolph, M. J. Barthel, U. Günther and F. H. Schacher, Polym. Chem., 2015, 6, 5633–5642 RSC.
- M. J. Barthel, T. Rudolph, A. Teichler, R. M. Paulus, J. Vitz, S. Hoeppener, M. D. Hager, F. H. Schacher and U. S. Schubert, Adv. Funct. Mater., 2013, 23, 4921–4932 CrossRef CAS.
- M. Wagner, M. J. Barthel, R. R. A. Freund, S. Hoeppener, A. Traeger, F. H. Schacher and U. S. Schubert, Polym. Chem., 2014, 5, 6943–6956 RSC.
- A. Zilkha and M. Weinstein, J. Appl. Polym. Sci., 1962, 6, 643–650 CrossRef CAS.
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed.
- V. Rejsek, D. Sauvanier, C. Billouard, P. Desbois, A. Deffieux and S. Carlotti, Macromolecules, 2007, 40, 6510–6514 CrossRef CAS.
- S. P. Carlotti, A. L. Labbé, V. Rejsek, S. P. Doutaz, M. Gervais and A. Deffieux, Macromolecules, 2008, 41, 7058–7062 CrossRef CAS.
- I. Kuntz, C. Cozewith, H. T. Oakley, G. Via, H. T. White and Z. W. Wilchinsky, Macromolecules, 1971, 4, 4–10 CrossRef CAS.
- I. Kuntz and W. R. Kroll, J. Polym. Sci., Part A: Polym. Chem., 1970, 8, 1601–1621 CrossRef CAS.
- H.-Q. Xie, J.-S. Guo, G.-Q. Yu and J. Zu, J. Appl. Polym. Sci., 2001, 80, 2446–2454 CrossRef CAS.
- Y. Kida, Y. Miura, K. Shikata and K. Azuma, J. Polym. Sci., Part A: Polym. Chem., 1975, 13, 2835–2842 CrossRef CAS.
- S. G. Reis, A. M. de Almeida, C. G. Diniz, V. L. da Silva and M. Le Hyaric, Chem. Biol. Drug Des., 2011, 78, 876–880 Search PubMed.
- The red color of the crosslinked FGE-containing material mainly came from DA cross-linked structure. The ECO/FGEm, BMI and ECO/FGEm/BMI0.5m were colorless, yellow and yellow, respectively, and there were no color changes when FGE and BMI were heated separately to 70 °C under a N2 atmosphere. Only when the mixture of FGE and BMI was heated to 70 °C, a time dependent color change from yellow to red can be observed during the heating procedure and a red DA product was obtained after the heating procedure.
- B. S. Beckingham, G. E. Sanoja and N. A. Lynd, Macromolecules, 2015, 48, 6922–6930 CrossRef CAS.
- M. R. Mason and A. M. Perkins, J. Organomet. Chem., 2000, 599, 200–207 CrossRef CAS.
- A. Keys, S. Bott and A. R. Barron, Chem. Commun., 1996, 20, 2339–2340 RSC.
- M. R. Mason, A. M. Perkins, R. M. Matthews, J. D. Fisher, M. S. Mashuta and A. Vij, Inorg. Chem., 1998, 37, 3734–3746 CrossRef CAS PubMed.
- Y. Chujo, K. Sada and T. Saegusa, Macromolecules, 1990, 23, 2636–2641 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, FT-IR spectra, GPC, DSC and TGA curves of representative polymer products. See DOI: 10.1039/c7py01516j |
|
| This journal is © The Royal Society of Chemistry 2018 |

 a,
Fang
Guo
*a,
Daohong
Liao
a,
Zhaomin
Hou
a,
Fang
Guo
*a,
Daohong
Liao
a,
Zhaomin
Hou








