
Search for a photoinduced (site-selective) cleavage of the Ar–Cl bond in dichloroanisoles†
Carlotta
Raviola
and
Maurizio
Fagnoni
 *
*
PhotoGreen Lab, Department of Chemistry, V. Le Taramelli 12, 27100 Pavia, Italy. E-mail: fagnoni@unipv.it; Fax: +39 0382 987323; Tel: +39 0382 987198
First published on 4th December 2017
Abstract
The site-selective cleavage of an Ar–X bond in polyhalogenated aromatics is an important tool in synthetic planning especially when more than one identical halogen atom is present. An alternative to the usual metal-catalyzed cleavage is represented by photochemistry although only a few examples have been reported. We then investigated the feasibility of the site-selective photodechlorination of some dichloroanisoles through a combined experimental and computational study. In the case of 2,4-dichloroanisole, selective detachment of the chlorine atom at the ortho position with respect to the OMe group was observed upon photohomolysis (in cyclohexane) or photoheterolysis (in MeOH) of the Ar–Cl bond. In the latter case, 5-chloro-2-methoxy-1,1′-biphenyl was exclusively formed upon reaction of the resulting phenyl cation with benzene. The substitution of an OH group for a OMe group was detrimental since a lower photoreactivity resulted with no improvement in the selectivity.
1. Introduction
Site-selective cleavage/activation of one halogen atom in dihaloarenes having the same halogen atoms is highly desirable even in view of their easy preparation. To this aim, metal-catalyzed procedures are the most diffuse1 but these lead in most cases to mixtures of mono- and disubstituted derivatives.2 Photochemistry may offer interesting (albeit rare) alternative routes for the selective cleavage of Ar–X bonds as shown in the selective hydrodefluorination of polyfluorinated aromatics by using Ir(ppy)3 as the photocatalyst.3 In recent years we deeply studied the behaviour of monohalogenated phenyl halides when irradiated under UV light.4–9 With aromatic chlorides (or fluorides) bearing an electron-donating group a photoheterolytic cleavage of the Ar–X bond took place with the concomitant formation of a phenyl cation.4–8 An intriguing example is that of the drug lomefloxacin (Scheme 1), where two fluorine atoms were present at positions 6 and 8 but only the Ar–F bond present at position 8 was selectively cleaved heterolytically upon UV irradiation.10 As for the above, we envisaged that related (simpler) dihaloaromatics having the same halogen atom could be used for a site-selective photochemical activation under metal-free conditions. In particular, we decided to focus our attention on dichloroanisoles.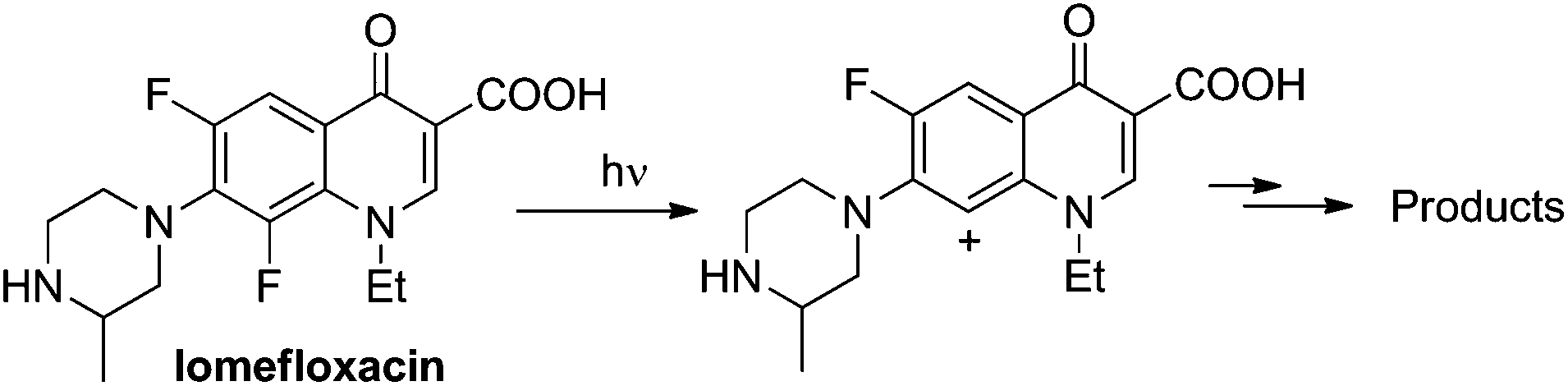 | ||
| Scheme 1 Photoinduced site-selective cleavage of an Ar–F bond. | ||
Only a few examples of photodehalogenation of dichloroanisoles have been reported in the literature. Irradiation of symmetrical 2,6-dichloroanisole and 3,5-dichloroanisole in methanol afforded monochlorinated products (2- and 3-dichloroanisoles, respectively) along with a modest amount of anisole.11,12 The case of unsymmetrical derivatives is more interesting. Indeed, the photolysis of 2,4-dichloroanisole and 2,3-dichloroanisole led to the selective cleavage of chlorine at the ortho position. In the former case the C2–Cl bond breakage was induced by a rhenium complex,13 while in the latter case simple irradiation in methanol led to a mixture of 3-chloroanisole and anisole.12
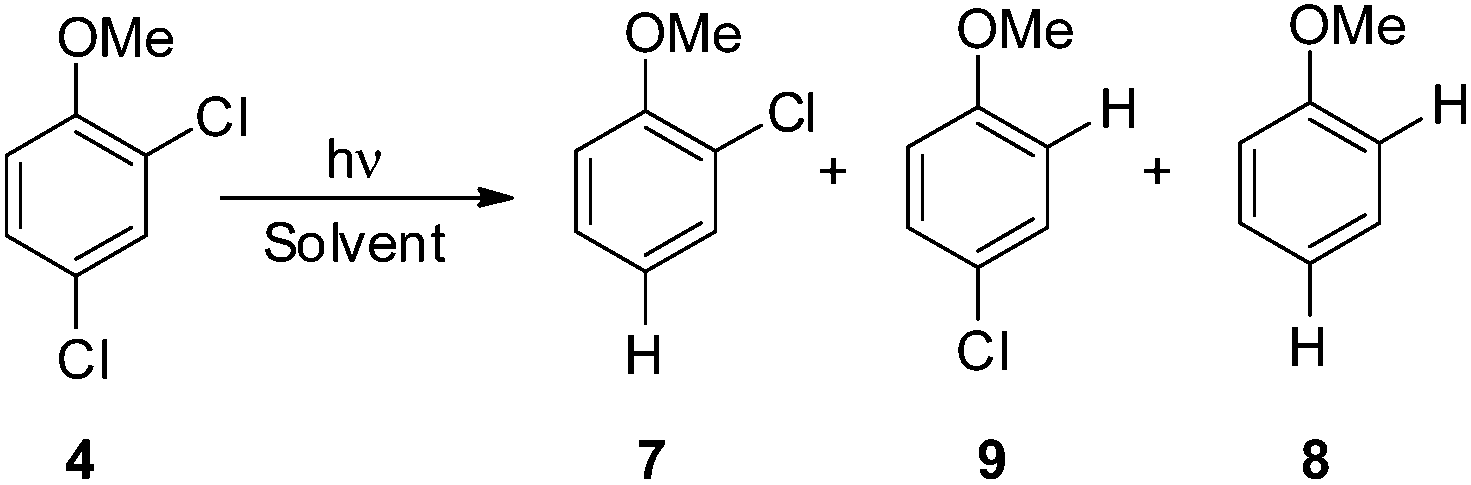
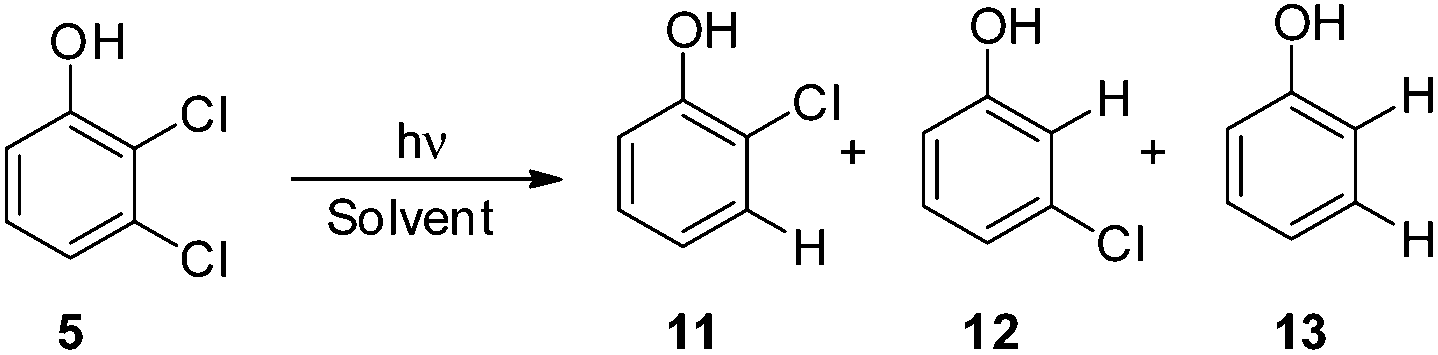
Thus, we decided to investigate in detail the photochemistry of all unsymmetrical dichloroanisoles (1–4, Scheme 2a). In particular, we studied their photochemical behavior in cyclohexane and MeOH upon irradiation at λ = 310 nm in order to assess whether a different selectivity may be observed when using two different photochemical activations. Indeed, we previously demonstrated that the photoinduced dechlorination of monochloroanisoles occurred from the first triplet excited state with two different mechanisms depending on the reaction solvent (homolysis in an apolar solvent and heterolysis in a polar one, Scheme 2b).8,14 This work was likewise supplemented by calculations aiming to rationalize the photoreactivity observed. Moreover, for the sake of comparison we also considered one dichlorophenol (5, Scheme 2a).
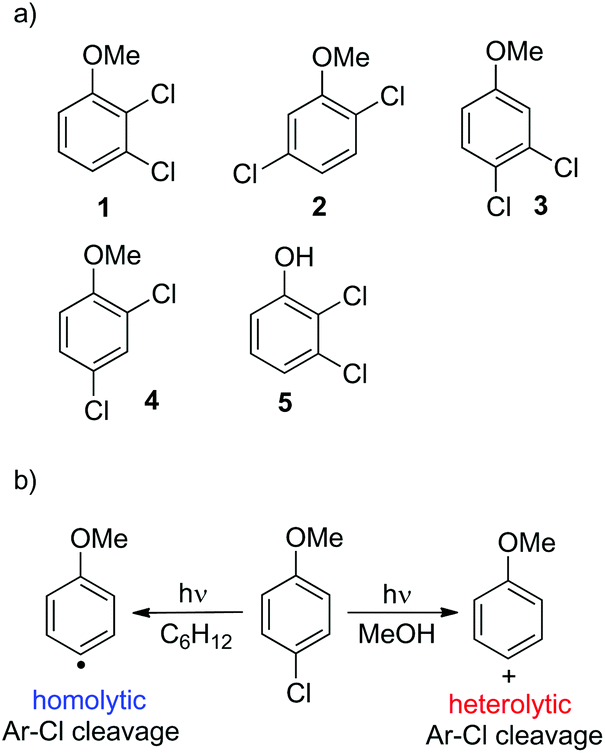 | ||
| Scheme 2 (a) Dichloroaromatics tested in this study and (b) the effect of medium on the photoinduced cleavage of the Ar–Cl bond in 4-chloroanisole. | ||
2. Experimental section
General
NMR spectra were recorded on 300 MHz spectrometer. The attributions were made on the basis of 1H and 13C NMR, as well as DEPT-135 experiments; chemical shifts are reported in ppm downfield from TMS. UV-Vis spectra were recorded with a double beam spectrophotometer equipped with a deuterium lamp (190–350 nm) and a halogen lamp (330–900 nm) and a photomultiplier R928. Fluorescence spectra were recorded with a luminescence spectrometer.Photochemical reactions were performed by using nitrogen-purged solutions in quartz tubes in a multilamp reactor fitted with ten 15 W phosphor coated lamps (emission centered at 310 nm) for irradiation. The reaction course was followed by GC analyses and the products formed were identified and quantified by comparison with authentic samples. Workup of the photolytes involved concentration in vacuo and chromatographic separation by using silica gel. Solvents of HPLC purity were employed in the photochemical reactions. Quantum yields were measured at 254 nm (1 Hg lamp, 15 W).
2,3-Dichloroanisole (1), 2,5-dichloroanisole (2), 3,4-dichloroanisole (3), 2,4-dichloroanisole (4), 2,3-dichlorophenol (5), 3-chloroanisole (6), 2-chloroanisole (7), anisole (8), 4-chloroanisole (9), 2-chlorophenol (11), 3-chlorophenol (12), phenol (13) and benzene are commercially available.
General procedure for the irradiation of dichloroaromatics 1–5
A 0.05 M solution of 1–5 in the chosen medium (MeOH, C6H12) was nitrogen purged in a quartz tube and irradiated in a multilamp reactor fitted with ten 15 W phosphor coated lamps (emission centered at 310 nm). The reaction course and the product distribution were monitored by GC analysis. The amounts of compounds 6–13 have been determined on the basis of calibration curves by comparison with commercial standards (see above).3. Results
Photophysical experiments were performed on 2,4-dichloroanisole (4) only, as a representative compound. The fluorescence quantum yields in cyclohexane (ΦF = 0.015) and in methanol (ΦF < 10−3) were very low (see Table S1† in the ESI).The decomposition quantum yield (Φ−1) of phenyl chlorides 1–5 (10−2 M) was determined by irradiation in cyclohexane and MeOH at 254 nm in comparison with monochlorinated anisoles (Table 1). As for 2- and 3-chloroanisoles, they are consistently more reactive in cyclohexane than in MeOH contrary to 4-chloroanisole where the photoreactivity was comparable (Φ−1ca. 0.3). The presence of an additional chlorine atom in dichloroanisoles did not change significantly the photoreactivity and the order in cyclohexane is 3 > 1 > 4 > 2 and in MeOH it is 2 > 3 > 1 ≈ 4. Again the photoreactivity of 1–4 was very high in apolar solvents and higher than in MeOH where it is low (<0.11), except for the case of 2. Changing a methoxy group with a hydroxy group dramatically decreased the reactivity in cyclohexane and to a lower extent in MeOH (compare the values of Φ−1 for 1 and 5).
| Substrate | Decomposition quantum yields (Φ−1)a | |
|---|---|---|
| Cyclohexane | MeOH | |
| a Measured at 254 nm in a 10−2 M solution. The values of the monochlorinated anisoles were reported for the sake of comparison. | ||
| 2-Chloroanisole | 0.74 | 0.11 |
| 3-Chloroanisole | 0.70 | 0.30 |
| 4-Chloroanisole | 0.26 | 0.26 |
| 1 | 0.57 | 0.09 |
| 2 | 0.39 | 0.29 |
| 3 | 0.69 | 0.11 |
| 4 | 0.48 | 0.08 |
| 5 | 0.11 | 0.03 |
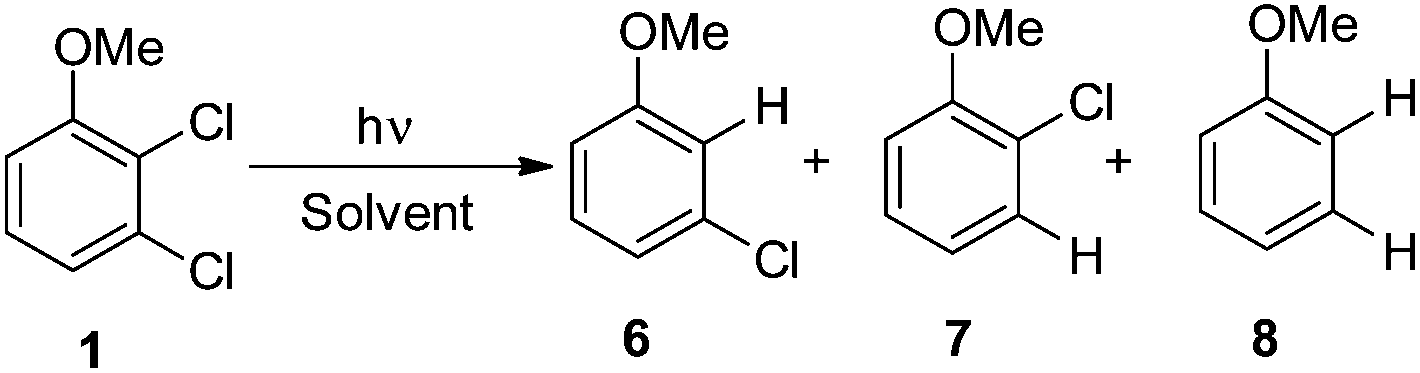
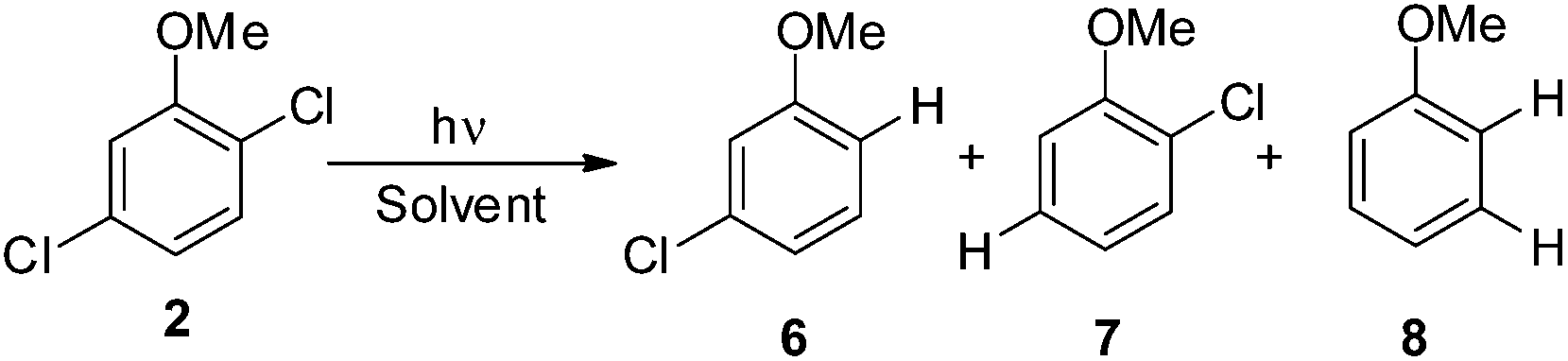
Irradiation experiments were carried out in cyclohexane and in methanol to induce (in analogy with 4-chloroanisole) a homolytic and a heterolytic cleavage of the Ar–Cl bond, respectively.8 No consumption of derivatives 1–5 was observed when irradiating the solutions covered with aluminum foil. 2,3-Dichloroanisole (1) was the first compound tested (Table 2) and irradiation of 1 in cyclohexane gave roughly an equimolar mixture of 3-chloroanisole (6) and 2-chloroanisole (7). Partial dechlorination of 6/7 to give anisole 8 occurred after 90 min of irradiation. GC analyses of the photolysate showed the presence of variable amounts of 1,1′-bi(cyclohexane) and chlorocyclohexane. On shifting to methanol, the reaction became more sluggish (64% consumption of 1 after 14 h) but again no selective ortho/meta dechlorination took place (Table 2).
|
|
||||
|---|---|---|---|---|
| Solvent | t irr (conversion %) | Products (yield %)b,c | ||
| 6 | 7 | 8 | ||
| a Irradiation carried out at λ = 310 nm with ten 15 W phosphor coated lamps on a 0.05 M solution of the dichloroaromatic in the solvent chosen. b Yields were determined by GC analysis. c In the experiments performed in cyclohexane, variable amounts of 1,1′-bi(cyclohexane) and chlorocyclohexane were detected by GC analysis. | ||||
| Cyclohexane | 15 min (18) | 9 | tr. | — |
| Cyclohexane | 30 min (39) | 15 | 14 | — |
| Cyclohexane | 60 min (55) | 20 | 16 | — |
| Cyclohexane | 90 min (79) | 27 | 21 | 8 |
| MeOH | 3 h (16) | 5 | 6 | — |
| MeOH | 7 h (32) | 11 | 12 | — |
| MeOH | 14 h (64) | 15 | 17 | 13 |
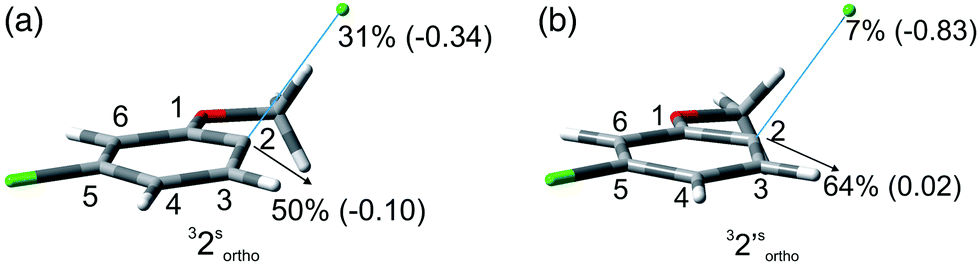
The experiments were supplemented by computational studies with the aim to rationalize the site-selectivity of the photoreaction. DFT (Density Functional Theory) at the M06-2X/def2TZVP level of theory was adopted to optimize the relevant structures as in previous studies on related chloroaromatics.9 Solvent effects (C6H12 and MeOH bulk) were included at the same level of theory adopting the SMD model (see the ESI† for further details).16 The photodehalogenation was supposed to arise from the triplet state since Intersystem Crossing (ISC) is very efficient in aromatic halides.17–19 We then considered for our purpose the most stable triplets in apolar or polar solvents. In the case of 31, calculations showed that in cyclohexane (or in methanol) the absolute minimum is 31ortho (or 31′ortho in MeOH) with the chlorine atom in position 2 out of the plane of the aromatic ring (see Fig. S2 and S3 in the ESI†). The non-planarity of the triplet state is in fair agreement with previous findings on (hetero)aromatic monohalides.9,17 The corresponding 31meta/31′meta isomers (see Fig. S4 and S5†) were less stable by about 6 kcal mol−1 (see the data collected in Table 7). Thus, calculations showed that in both apolar and polar solvents there is a preference for the cleavage of the ortho Ar–Cl bond (Ar–Clortho). In order to evaluate the energy required to detach the Clmeta atom in 31meta (ΔEmeta) and the Clortho atom in 31ortho (ΔEortho), the Ar–Cl bond was elongated up to 3 Å, a distance where the bond can be considered broken. Comparable values were obtained for the two isomers in cyclohexane (ca. 15 kcal mol−1, Table 7) consistently higher than those in methanol (the difference between ΔEmeta and ΔEortho was ca. 8–10 kcal mol−1).9 The stretched structures in cyclohexane (31sortho) and in MeOH (31′sortho) of the most stable conformers are shown in Fig. 1a and Fig. 1b, respectively.
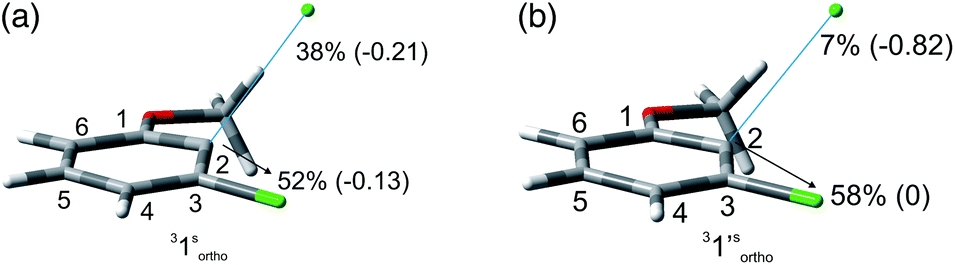 | ||
| Fig. 1 Geometries, spin densities and ESP charge (in parentheses) calculated in C6H12 (a) and MeOH (b) bulk at the SMD-M06-2X/def2TZVP level for 31sortho and 31′sortho upon stretching the Ar–Clortho bond up to 3 Å. | ||
The homolysis of the Ar–Cl bond in cyclohexane was confirmed since the spin density was almost equally localized on C2 (52%) and on the chlorine atom (38%, Fig. 1a). In contrast, the heterolysis in MeOH was witnessed by the significant negative charge developed at the chlorine atom (−0.82, Fig. 1b).
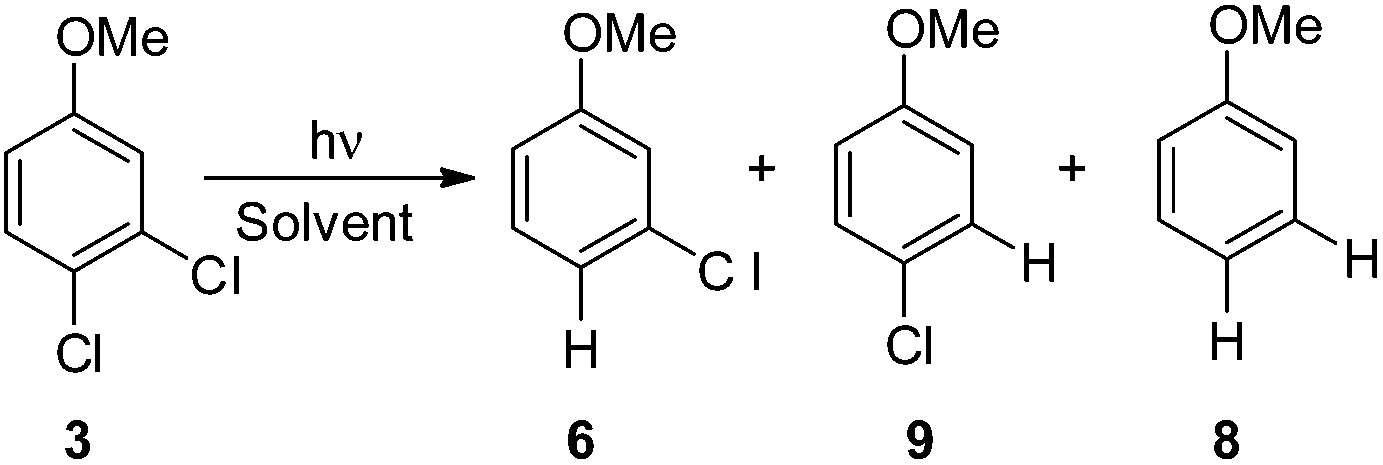
In order to investigate a different ortho/meta preference with respect to the OMe group in the photodechlorination, we then irradiated 2,5-dichloroanisole (2, Table 3). Interestingly, different from the previous case, irradiation both in cyclohexane and in MeOH led to a preference for the release of the chlorine atom placed at the ortho position (6/7ca. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in the first case and ca. 3–4:1 in the second case). Although the rate of consumption of product 2 in cyclohexane seemed comparable to that of 1, the consumption of compound 2 in methanol was much higher (83% in 6 h). Computational results were similar to those obtained for 1 (Table 7), showing again a preference for the cleavage of the Ar–Clortho bond (see the structures 32sortho and 32′sortho in Fig. 2 and Fig. S6–S9† for the other minima) confirming the homolysis in cyclohexane and heterolysis in MeOH.
:1 in the first case and ca. 3–4:1 in the second case). Although the rate of consumption of product 2 in cyclohexane seemed comparable to that of 1, the consumption of compound 2 in methanol was much higher (83% in 6 h). Computational results were similar to those obtained for 1 (Table 7), showing again a preference for the cleavage of the Ar–Clortho bond (see the structures 32sortho and 32′sortho in Fig. 2 and Fig. S6–S9† for the other minima) confirming the homolysis in cyclohexane and heterolysis in MeOH.
 | ||
| Fig. 2 Geometries, spin densities and ESP charge (in parentheses) calculated in C6H12 (a) and MeOH (b) bulk at the SMD-M06-2X/def2TZVP level for 32sortho and 32′sortho upon stretching the Ar–Clortho bond up to 3 Å. | ||
|
|
||||
|---|---|---|---|---|
| Solvent | t irr (conversion %) | Products (yield %)b,c | ||
| 6 | 7 | 8 | ||
| a Irradiation carried out at λ = 310 nm with ten 15 W phosphor coated lamps on a 0.05 M solution of the dichloroaromatic in the solvent chosen. b Yields were determined by GC analysis. c In the experiments performed in cyclohexane, variable amounts of 1,1′-bi(cyclohexane) and chlorocyclohexane were detected by GC analysis. | ||||
| Cyclohexane | 10 min (18) | 9 | tr. | — |
| Cyclohexane | 30 min (32) | 18 | 9 | — |
| Cyclohexane | 60 min (54) | 27 | 13 | — |
| Cyclohexane | 90 min (82) | 42 | 17 | 5 |
| Cyclohexane | 120 min (88) | 49 | 18 | 10 |
| Methanol | 1.5 h (36) | 29 | 6 | — |
| Methanol | 3 h (62) | 45 | 14 | — |
| Methanol | 6 h (83) | 60 | 17 | 5 |
The photochemistry of 3,4-dichloroanisole (3) gave an indication about the meta/para selectivity (Table 4). In cyclohexane, an almost complete consumption of 3 was achieved after 90 min. In this case the cleavage of the meta chlorine atom was preferred and, interestingly, no complete dechlorination took place even at such high conversion (ca. 95%). In methanol, despite the sluggish conversion, no product 6 was formed and the selectivity was complete for the meta position albeit a strong competitive further dechlorination operated here to form 8. Also in this case two minima were found: one with the meta chlorine atom (33meta) and the other with the para chlorine atom (33para) out of the plane of the aromatic ring. Unfortunately, these differed only by about 1 kcal mol−1 (Table 7) and so calculations were not able to give an indication on the selectivity of the reaction, although the Ar–Clpara bond was slightly preferred (see Fig. 3 and Fig. S10–13† for the other structures). As already reported for compounds 1 and 2, the energy calculated to detach the chlorine atom is higher in apolar solvents than in polar solvents (Table 7).
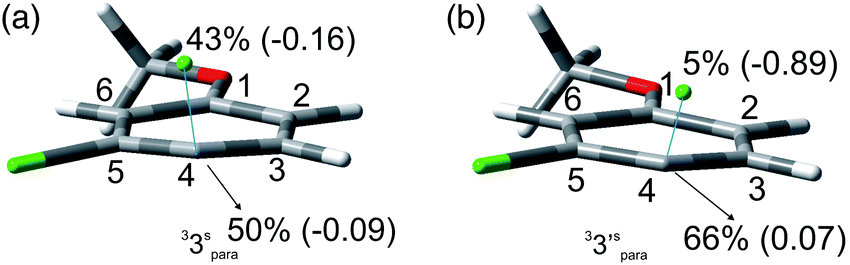 | ||
| Fig. 3 Geometries, spin densities and ESP charge (in parentheses) calculated in C6H12 (a) and MeOH (b) bulk at the SMD-M06-2X/def2TZVP level for 33spara and 33′spara upon stretching the Ar–Clpara bond up to 3 Å. | ||
|
|
||||
|---|---|---|---|---|
| Solvent | t irr (conversion %) | Products (yield %)b,c | ||
| 6 | 9 | 8 | ||
| a Irradiation carried out at λ = 310 nm with ten 15 W phosphor coated lamps on a 0.05 M solution of the dichloroaromatic in the solvent chosen. b Yields were determined by GC analysis. c In the experiments performed in cyclohexane, variable amounts of 1,1′-bi(cyclohexane) and chlorocyclohexane were detected by GC analysis. | ||||
| Cyclohexane | 5 min (14) | — | 8 | — |
| Cyclohexane | 10 min (27) | — | 13 | — |
| Cyclohexane | 15 min (34) | 5 | 17 | — |
| Cyclohexane | 30 min (43) | 5 | 21 | — |
| Cyclohexane | 60 min (78) | 8 | 34 | — |
| Cyclohexane | 90 min (95) | 12 | 49 | — |
| Methanol | 21 h (39) | — | 31 | 8 |
| Methanol | 32 h (50) | — | 25 | 25 |
| Methanol | 48 h (60) | — | 18 | 29 |
| Methanol | 62 h (64) | — | 16 | 35 |
We then reasoned that since an ortho vs. meta preference was found in compound 2 and a meta vs. para preference was observed in derivative 3, a marked preference in the Ar–Cl cleavage from the ortho position in ortho/para dichlorinated anisole should be expected. This led us to consider 2,4-dichloroanisole (4) as a promising substrate for a site-selective cleavage. Interestingly, in cyclohexane, 4-chloroanisole (9) is the only photoproduct formed up to 79% conversion of 4 (Table 5). Further irradiation caused the conversion of 9 to anisole. In methanol, the reaction was still selective, albeit sluggish, and with a poor mass balance. In accordance with experimental data, calculations demonstrated both in cyclohexane and in methanol that the absolute minimum was that causing the Ar–Clortho cleavage (see Fig. 4 and Fig. S14–S17† for the other structures). The energy barrier for the Ar–Cl cleavage was similar in 34spara and 34sortho and higher in cyclohexane (ΔE ≈ 15–18 kcal mol−1) rather than in methanol (ΔE ≈ 5–7 kcal mol−1, Table 7).
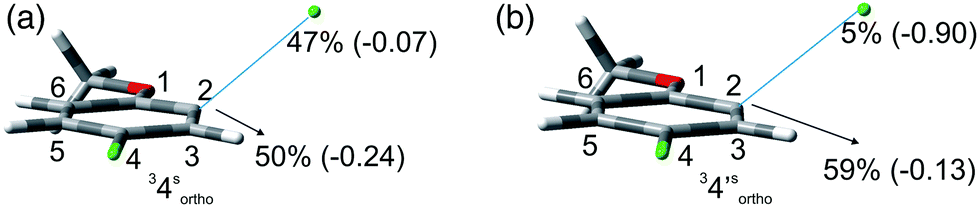 | ||
| Fig. 4 Geometries, spin densities and ESP charge (in parentheses) calculated in C6H12 (a) and MeOH (b) bulk at the SMD- M06-2X/def2TZVP level for 34sortho and 34′sortho upon stretching the Ar–Clortho bond up to 3 Å. | ||
|
|
||||
|---|---|---|---|---|
| Solvent | t irr (conversion %) | Products (yield %)b,c | ||
| 7 | 9 | 8 | ||
| a Irradiation carried out at λ = 310 nm with ten 15 W phosphor coated lamps on a 0.05 M solution of the dichloroaromatic in the solvent chosen. b Yields were determined by GC analysis. c In the experiments performed in cyclohexane, variable amounts of 1,1′-bi(cyclohexane) and chlorocyclohexane were detected by GC analysis. | ||||
| Cyclohexane | 5 min (6) | — | 6 | — |
| Cyclohexane | 10 min (30) | — | 13 | — |
| Cyclohexane | 30 min (49) | — | 26 | — |
| Cyclohexane | 60 min (79) | — | 40 | — |
| Cyclohexane | 90 min (95) | — | 57 | 5 |
| Cyclohexane | 120 min (100) | — | 59 | 18 |
| Cyclohexane | 150 min (100) | — | 37 | 35 |
| Methanol | 8 h (37) | — | 15 | — |
| Methanol | 16 h (54) | — | 22 | — |
| Methanol | 24 h (63) | — | 25 | — |
| Methanol | 46 h (70) | — | 26 | 12 |
Having these positive results on 4, we tried to perform a selective arylation at the ortho position by photoheterolytic cleavage in polar solvents by using benzene as the nucleophile. Since the reaction is quite sluggish in MeOH and this solvent is known to efficiently react with phenyl cations by hydrogen atom transfer,8 we shifted to 2,2,2-trifluoroethanol (TFE) in the presence of acetone known to accelerate the conversion of aryl chlorides by energy transfer.14 The conversion was kept low in order to avoid competitive dechlorination of the resulting arylated product. Gratifyingly, photolysis of 4 in TFE in the presence of benzene (2 M) after 9 h cleanly gave methoxybiphenyl 10 in 45% yield (Scheme 3).
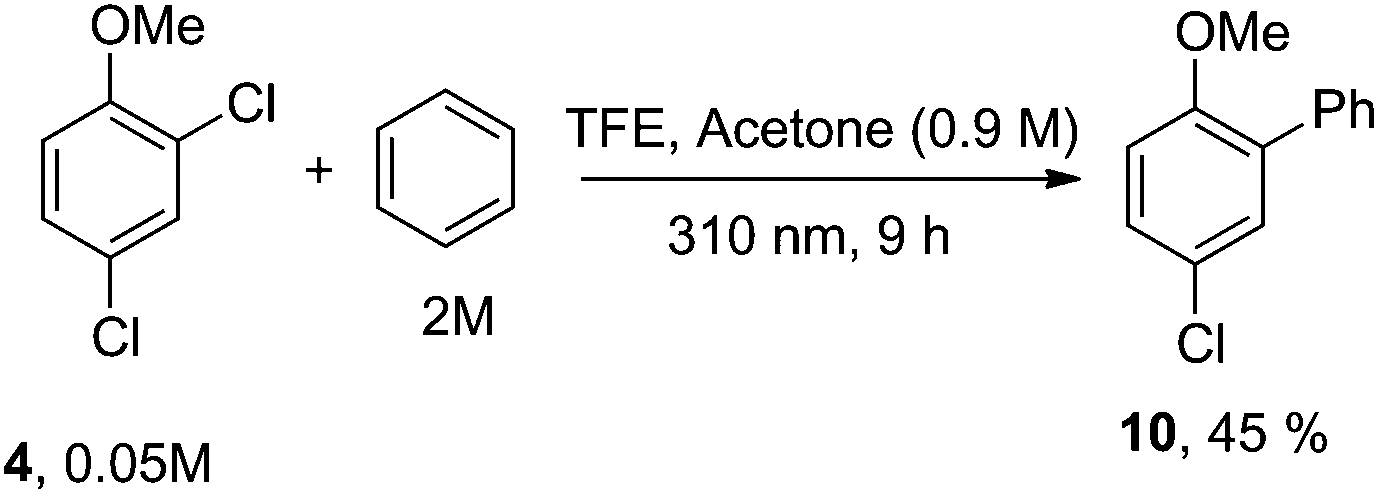 | ||
| Scheme 3 Selective metal-free arylation of 2,4-dichloroanisole (4). Isolated yield based on 31% consumption of 4. | ||
As the last substrate tested, we considered 2,3-dichlorophenol (5) in order to see whether a free OH group may exert a positive effect on the site selective C–Cl cleavage with respect to the corresponding anisole 1. Disappointingly, the reactions required a long time to occur (up to 48 h) and in cyclohexane the reaction did not reach completion (Table 6). In both cases the observed selectivity was not so different from that observed in the case of 1 and a low mass balance was obtained in the case of cyclohexane. As already observed for compound 1 we found two minima: 35meta and the more stable 35ortho with the meta and ortho chlorine atoms out of the plane, respectively. Noteworthily, both in cyclohexane and methanol the shift from 1 to 5 slightly affected the energy barrier (see Fig. 5 and Table 7, Fig. S18–S21†).
 | ||
| Fig. 5 Geometries, spin densities and ESP charge (in parentheses) calculated in C6H12 (a) and MeOH (b) bulk at the SMD-M06-2X/def2TZVP level for 35sortho and 35′sortho upon stretching the Ar–Clortho bond up to 3 Å. | ||
|
|
||||
|---|---|---|---|---|
| Solvent | t irr (conversion %) | Products (yield %)b,c | ||
| 11 | 12 | 13 | ||
| a Irradiation carried out at λ = 310 nm with ten 15 W phosphor coated lamps on a 0.05 M solution of the dichloroaromatic in the solvent chosen. b Yields were determined by GC analysis. c In the experiments performed in cyclohexane, variable amounts of 1,1′-bi(cyclohexane) and chlorocyclohexane were detected by GC analysis. | ||||
| Cyclohexane | 6 h (30) | 6 | 4 | — |
| Cyclohexane | 14 h (63) | 5 | 3 | 5 |
| Cyclohexane | 24 h (65) | 4 | 4 | 9 |
| Methanol | 24 h (14) | 4 | 10 | — |
| Methanol | 48 h (100) | 22 | 25 | — |
| Structures | Solvent | ΔEconfa (kcal mol−1) |
ΔEC–Clxb(kcal mol−1) |
Spin density (ESP charge) at Clx atomc | Spin density (ESP charge) at Clx atomd |
|---|---|---|---|---|---|
| a Energy difference between the two conformers. The most stable conformers are given in bold. b Energy required to stretch the Ar–Clx bond up to 3 Å. c At equilibrium. d At 3 Å. | |||||
| 31meta | C6H12 | 5.7 | Clmeta = 15.6 | 8% (−0.12) | 49% (−0.02) |
| 31ortho | C6H12 | Cl ortho = 14.8 | 9% (−0.11) | 38% (−0.21) | |
| 3 1 ′ meta | MeOH | 5.9 | Clmeta = 10.6 | 8% (−0.15) | 4% (−0.91) |
| 3 1 ′ ortho | MeOH | Cl ortho = 8.1 | 10% (−0.19) | 7% (−0.82) | |
| 3 2 meta | C6H12 | 5.8 | Clmeta = 14.5 | 7% (−0.13) | 48% (−0.05) |
| 3 2 ortho | C6H12 | Cl ortho = 13.7 | 9% (−0.13) | 50% (−0.10) | |
| 3 2 ′ meta | MeOH | 5.6 | Clmeta = 3.9 | 7% (−0.16) | 4% (−0.91) |
| 3 2 ′ ortho | MeOH | Cl ortho = 6.6 | 9% (−0.20) | 7% (−0.83) | |
| 3 3 meta | C6H12 | 0.6 | Clmeta = 17.6 | 7% (−0.12) | 50% (−0.02) |
| 3 3 para | C6H12 | Cl para = 17.7 | 8% (−0.14) | 43% (−0.16) | |
| 3 3 ′ meta | MeOH | 1.1 | Clmeta = 7.3 | 7% (−0.16) | 4% (−0.91) |
| 3 3 ′ para | MeOH | Cl para = 7.5 | 8% (−0.21) | 5% (−0.89) | |
| 3 4 para | C6H12 | 2.6 | Clpara = 15.6 | 8% (−0.15) | 44% (−0.14) |
| 3 4 ortho | C6H12 | Cl ortho = 18.7 | 8% (−0.10) | 47% (−0.07) | |
| 3 4 ′ para | MeOH | 2.1 | Clpara = 5.0 | 8% (−0.21) | 5% (−0.90) |
| 3 4 ′ ortho | MeOH | Cl ortho = 7.2 | 8% (−0.15) | 5% (−0.90) | |
| 3 5 meta | C6H12 | 2.8 | Clmeta = 14.9 | 8% (−0.12) | 49% (−0.02) |
| 3 5 ortho | C6H12 | Cl ortho = 17.1 | 9% (−0.11) | 43% (−0.14) | |
| 3 5 ′ meta | MeOH | 3.6 | Clmeta = 8.8 | 8% (−0.17) | 4% (−0.90) |
| 3 5 ′ ortho | MeOH | Cl ortho = 8.7 | 10% (−0.19) | 6% (−0.87) | |
As for the photoheterolytic cleavage in compounds 1–4 a further computational study was performed on selected phenyl cations. In accordance with 2-methoxyphenyl cations (1/319+), 314+, 315+, and 317+ showed a ring with a geometry close to a regular hexagon, while 115+ was planar with a cumulative double bond character at the C1–C2–C3 moiety (see Fig. 6b and Fig. S28–S31†). In contrast, 114+ and 117+ (see Fig. 6a and Fig. S29, S31†) were characterized by a puckering of the ring and a small out of plane displacement. For completeness we have also investigated the geometry of the corresponding radical 24˙ that was found to be planar (Fig. 6c and Fig. S32, S33†).
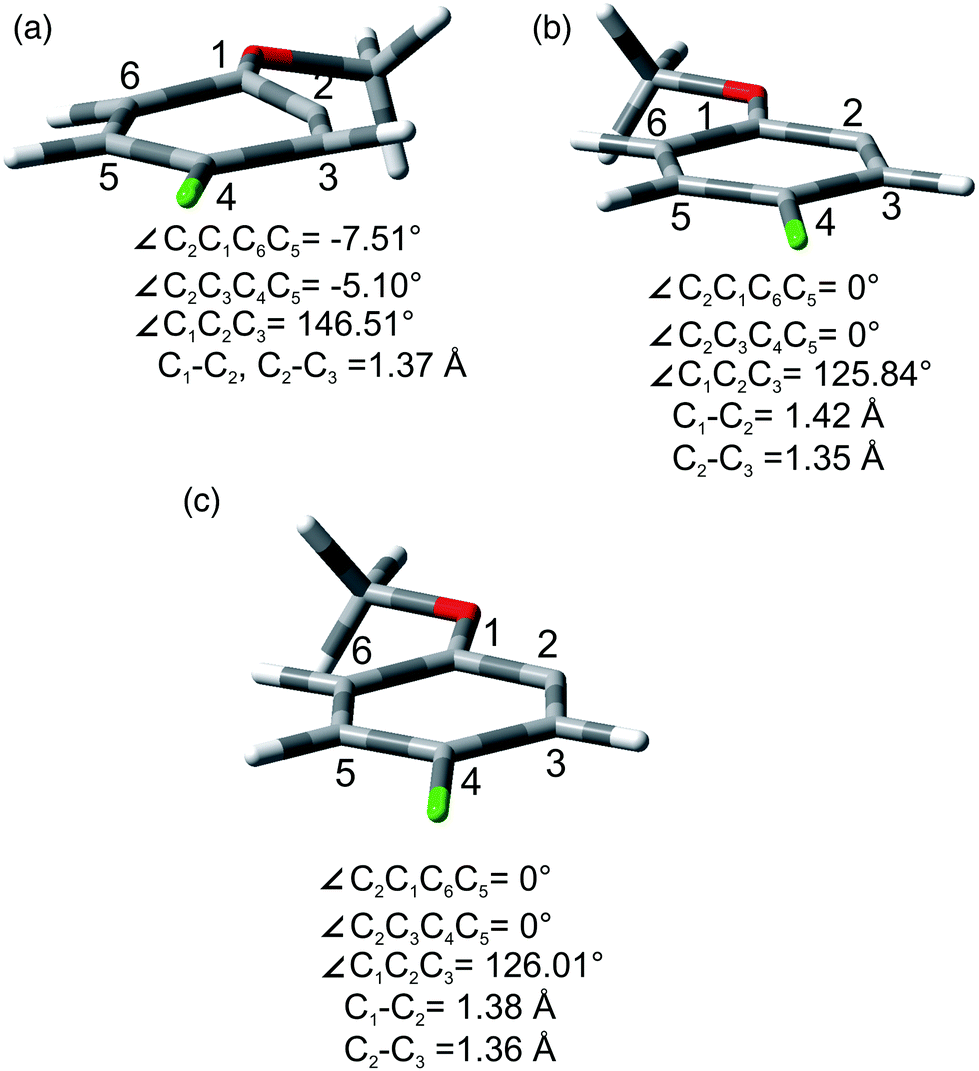 | ||
| Fig. 6 Bond lengths (Å), angles (∠, in degrees) for 117+ (a), 317+ (b), and 24 (c). | ||
In particular, the isodesmic reaction reported in eqn (1) was adopted in solution (MeOH) to evaluate the (de)stabilization imparted by the methoxy group and chlorine atom with respect to the parent singlet phenyl cation (1Ph+, Fig. 7). The presence of an ortho-methoxy group on the parent phenyl cation (see cations 1,319+) exerted a great stabilization on the corresponding triplet 319+ and a slight destabilization on the singlet 119+. The chlorine atom, however, exerted a destabilization effect on cations 1,319+ (less than 8 kcal mol−1 for triplets and in the 7–17 kcal mol−1 range for singlets). The most important effect was observed in cation 14+ when the chlorine atom is adjacent to the cationic site (Fig. 7). At any rate, in each case the energy gap between the singlet and the triplet states was increased by the presence of the chlorine atom, the triplets being again the most stable cations.
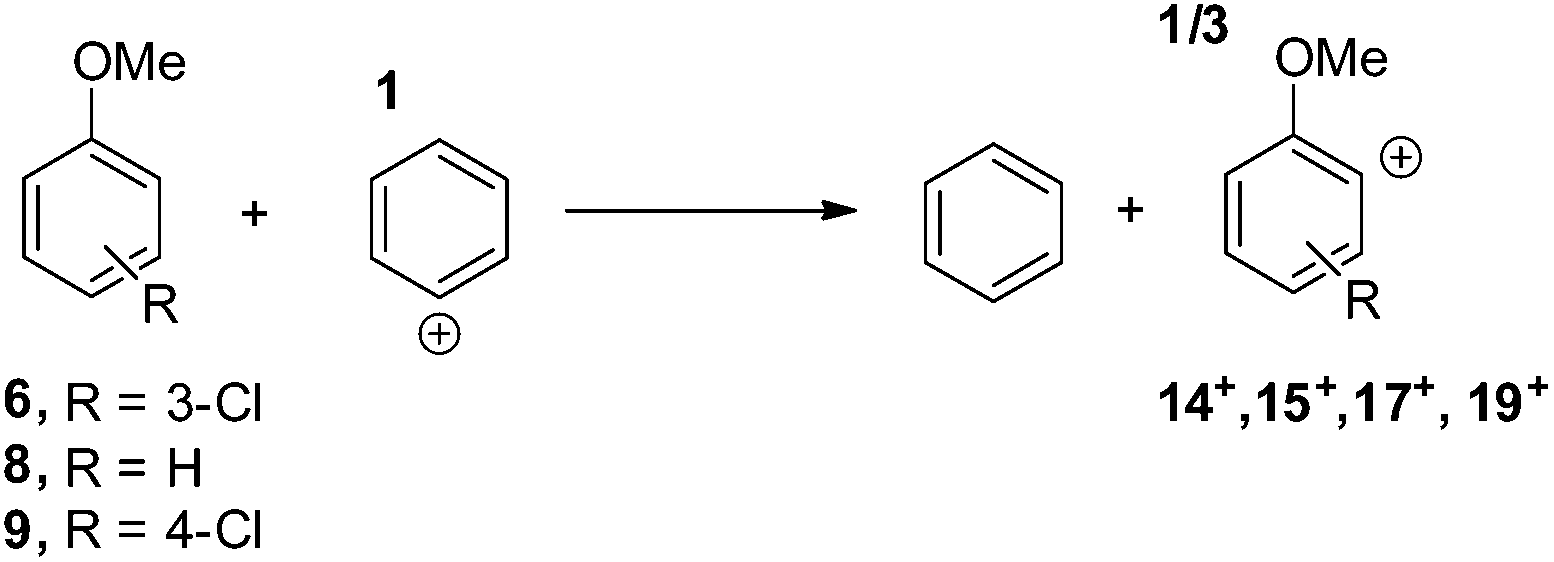 | (1) |
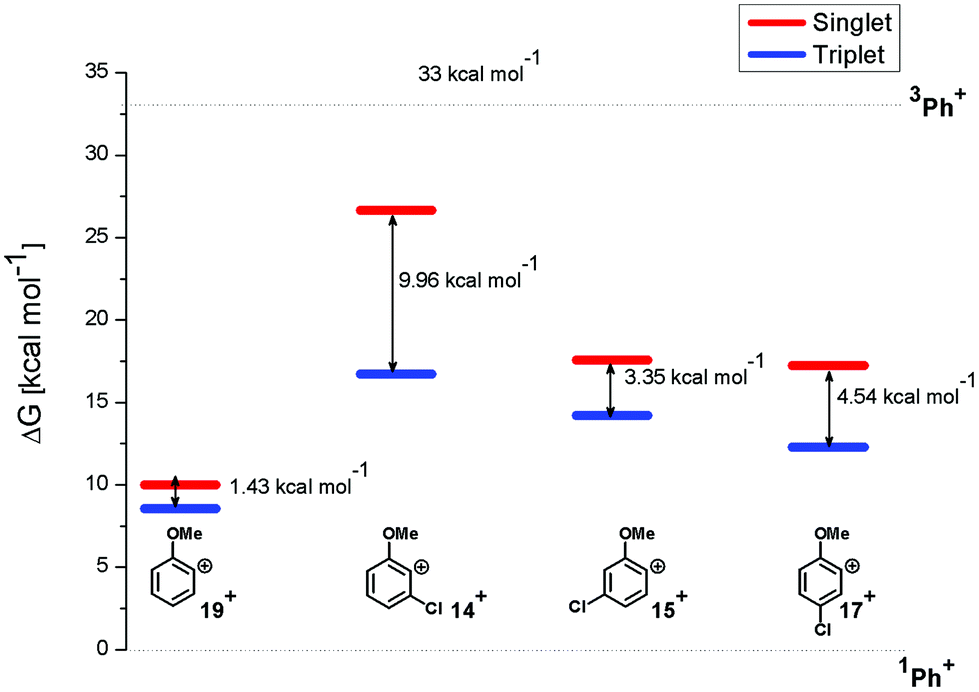 | ||
| Fig. 7 Relative Gibbs free energies (see Tables S2 and S3 in the ESI†) of singlet (red) and triplet (blue) phenyl cations 14+, 15+, 17+, and 19+ in MeOH solution according to isodesmic reaction reported in eqn (1). | ||
For the sake of comparison, a computational investigation on the photohomolytic cleavage by forming the corresponding phenyl radicals was likewise carried out. The isodesmic eqn (2) was used in solution (C6H12) to evaluate the (de)stabilization imparted by the methoxy group and chlorine atom with respect to the parent phenyl radical (Ph˙, Fig. 8).
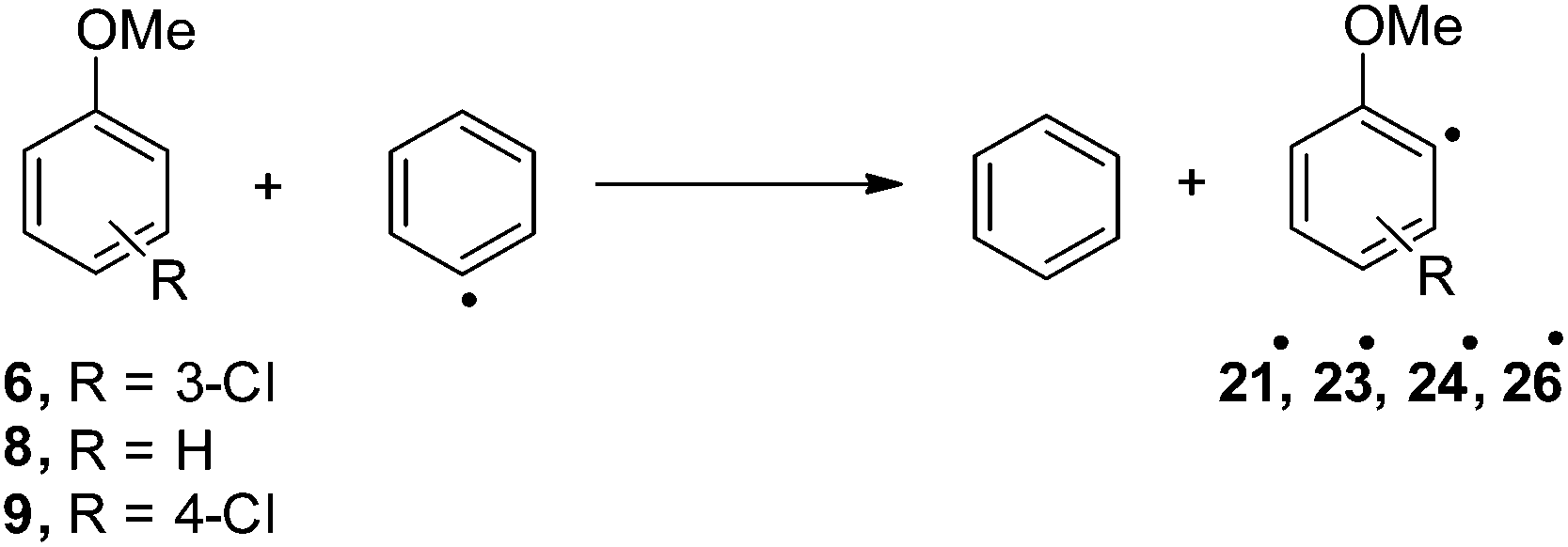 | (2) |
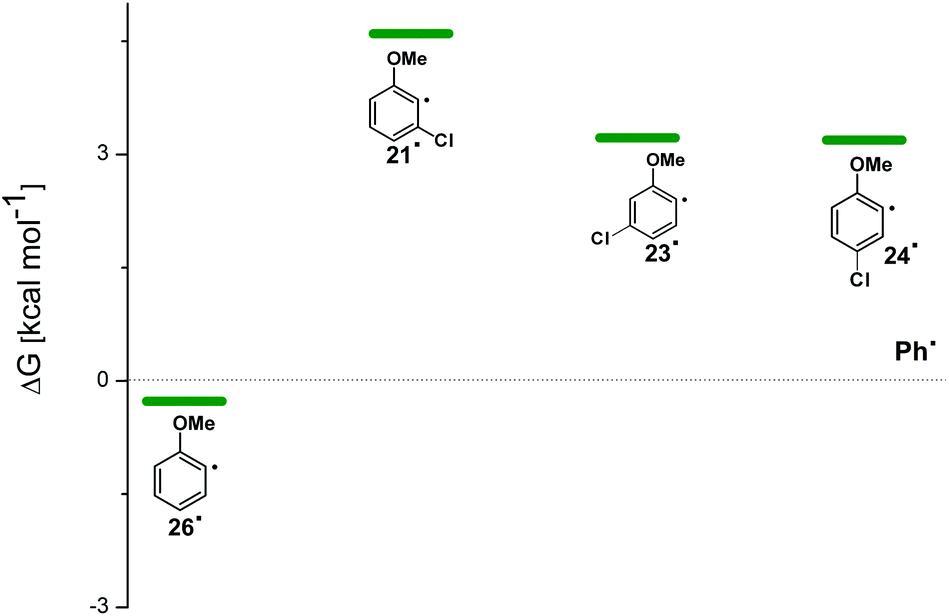 | ||
| Fig. 8 Relative Gibbs free energies (see Tables S4 and S5†) of phenyl radicals 21˙, 23˙, 24˙, and 26˙ in C6H12 solution according to isodesmic reaction reported in eqn (2). | ||
4. Discussion
Photoreactivity of compounds 1–5
Compounds 1–4 underwent chlorine loss upon irradiation both in apolar and polar solvents and in all cases the efficiency in cyclohexane (Φ−1 up to 0.7) was higher than that in MeOH (Φ−1 < 0.3), roughly following the trend shown for monochlorinated anisoles. As for the last point, the presence of a further chlorine atom in 1–4 did not significantly interfere with their photoreactivity. Quite surprisingly, dichlorophenol 5 was the less photoreactive compound tested both in cyclohexane and MeOH. Here the effect of the presence of a further chlorine atom was apparent, since 4-chlorophenol in MeOH was by far more photoreactive than 4-chloroanisole.21 The energy calculated to cleave the Ar–Cl bond (ΔE) is consistently higher in cyclohexane than in MeOH although the process is more efficient in the former case. The same trend resulted from the calculations carried out on monochlorinated anisoles (see Fig. S22–S27†). A tentative mechanism is reported in Scheme 4. Irradiation of compounds 1–5 in polar solvents led to a heterolytic cleavage of one Ar–Cl bond to form the corresponding triplet cations 314–18+ (path a). Hydrogen abstraction from methanol led to the corresponding monochlorinated compounds 6, 7, 9, 11, and 12 (path b).6,7 Addition of a nucleophile (benzene) and shifting to a high stabilizing ion solvent (TFE) allowed for the formation of biphenyl 10via selective Ar–Cl cleavage in compound 4 (path c). The reaction with benzene is diagnostic of the presence of a triplet phenyl cation.6,7 Different from thermal reactions (see below) no disubstitution products were formed (even in traces) under our conditions despite the presence of a large amount of the nucleophile. The absence of photosolvolysis products is an indication of the lack of ISC from cations 314–18+ to 114–18+ (path d). This is in accord with the isodesmic reaction reported in eqn (1) for cations 14+, 15+, and 17+ (Fig. 7) that clearly showed that the lower lying state of the cations is the triplet. This is in contrast to what was found for 4-chloroanisole where 1,4-dimethoxybenzene was obtained upon irradiation in MeOH.8 This is again an indication of the great effects that the position and the nature of the substituents, present on the phenyl cation ring, exert on the relative stability of singlet and triplet phenyl cations. As apparent from Fig. 7, the presence of a chlorine atom (in the role of a σ acceptor) in cations 14+, 15+, and 17+ induced a destabilization both on the singlets and on the triplets (more pronounced in the former case). This effect is enhanced when the chlorine atom is close to the cationic site (cation 14+) where the S–T gap is around 10 kcal mol−1 and the singlet is destabilized by ca. 17 kcal mol−1 with respect to the singlet 2-methoxyphenyl cation 119+. The concomitant effect of the methoxy group and the chlorine atom, however, is sufficient to make triplets (and not singlets) by far more stable than the corresponding parent triplet phenyl cation 3Ph+ (Fig. 7). In an apolar solvent a photohomolysis took place generating the corresponding aryl radicals (path e) that easily abstract hydrogen from cyclohexane (path f) to give again monochlorinated anisoles and phenols. The concomitant generation of the cyclohexyl radical was responsible for the formation of chlorocyclohexane and bicyclohexyl byproducts (paths f′ and f′′). The presence of the latter compounds is diagnostic of the involvement of an aryl radical in the process. In this case the presence of an ortho-methoxy group slightly stabilized the parent phenyl radical but addition of a further chlorine atom imparted a destabilization of 3–5 kcal mol−1. The high photoreactivity of monochlorinated anisoles (phenols) (see Table 1) in both of the solvents tested led to a further photodechlorination to anisole 8 or phenol 13 (Scheme 4, path g).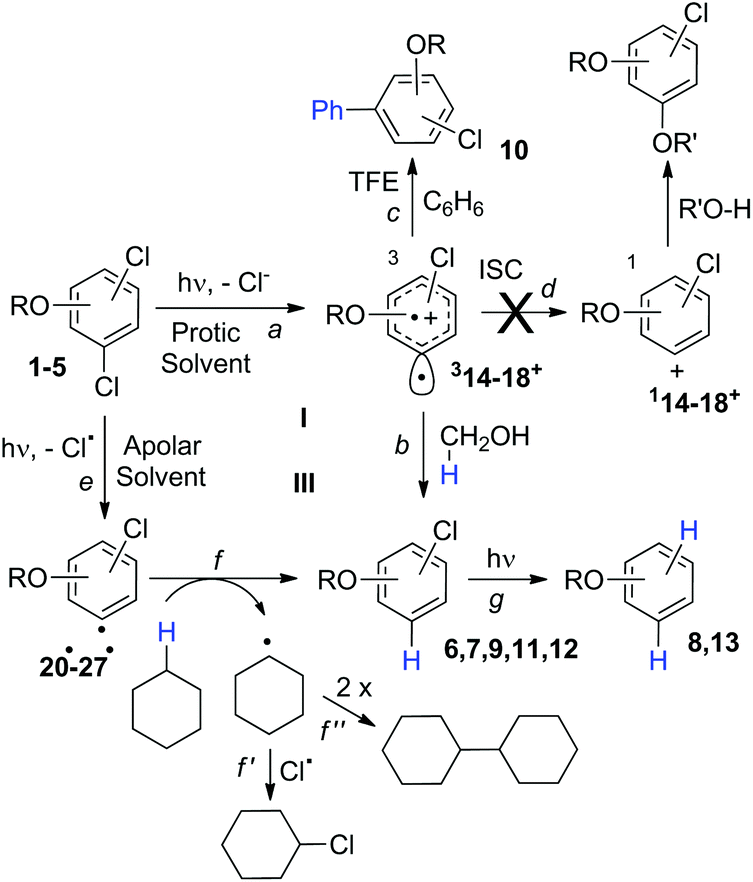 | ||
| Scheme 4 Proposed mechanism. | ||
Computational data confirmed that even in dichloroanisoles and phenols the cleavage of the Ar–Cl bond takes place from the triplet state and it is homolytic in cyclohexane whereas it is heterolytic in methanol. Taking 2,4-dichloroanisole (4) as a model, both in C6H12 and in MeOH the C2 atom sticks out of the plane of the aromatic ring at the equilibrium and becomes planar after stretching of the Ar–Clortho bond up to 3 Å (see Fig. 4 and Fig. S14–S17†).
The elongation of the Ar–Clortho bond in cyclohexane resulted in a non-significant charge formation on the chlorine atom (−0.07) and the spin distribution is mainly localized at C2 (50%) and Cl atoms (47%) fully supporting the homolysis. In methanol, a marked charge separation (Cl −0.90) is apparent with a concomitant low spin localization on the chlorine atom (5%) in accordance with the formation of a triplet cation.
Site-selective Ar–Cl bond activation
The site-selective cleavage of the Ar–Cl bond in the compounds examined is a difficult task. Indeed, the calculated bond dissociation energies (BDE) (for further details see section 2.1 in the ESI†) of the Ar–Cl bonds in compounds 1 and 4 showed that there is no significant difference between Ar–Clortho and Ar–Clmeta in the former case and between Ar–Clortho and Ar–Clpara in the latter (Fig. 9). As expected, the compounds have a planar geometry in marked contrast to their triplet excited states (Fig. 1 and 4).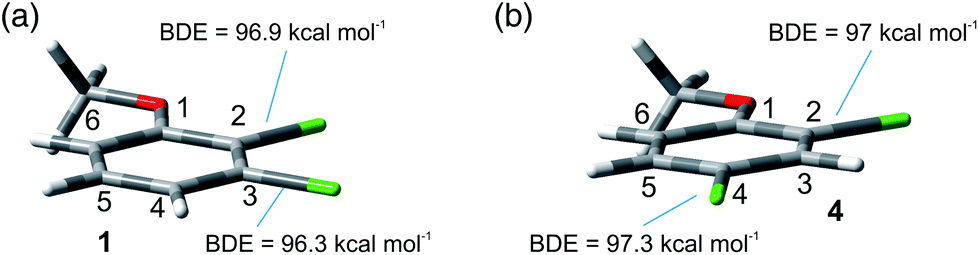 | ||
| Fig. 9 Geometries and BDE calculated in gas at the M06-2X/def2TZVP level for 1 and 4. | ||
For the sake of comparison, we analyzed what is so far reported for the activation of the Ar–Cl bonds in compounds 1–5. The data are collected in Table 8. Five methods were described for the tentative selective cleavage of the Ar–Cl bond in compound 1 giving contrasting results. In fact, under Pd catalysis the reaction with a strong (PhMgBr, entry 1) or a weak (an aniline, entries 2 and 3) nucleophile gave preferentially the substitution at the 3 position in the former case and at the 2 position in the latter case. Moreover, disubstitution is an important competitive path (see again entry 1). Copper catalysis led to a complete disubstitution pattern in the reaction with Me3SiCl/Mg (entry 4) whereas in Ni catalyzed reaction with EtMgBr an almost exclusive preference for the 3 position resulted (entry 5). Pd catalyzed addition of PhB(OH)2 onto 1 was not selective leading to disubstitution (entry 6). Only one example was reported on the reactivity of compound 2 where Pd catalysis has a preference for the activation of the chlorine at position 3 (entry 7). Similarly, a poorly selective functionalization was found in compound 3 (entries 8–11, Table 8) in some cases depending on the nucleophile used (compare entries 9 and 10). Different conditions were tested in the Ar–Cl cleavage in compound 4 (entries 12–16). Here the disubstitution is a serious drawback even under electrolytic dechlorination (entry 14). Nevertheless, a complete regioselectivity at position 2 was reported in the cobalt catalyzed carboxylation (entry 15) and under irradiation in the presence of Cp*Re(CO)3 (entry 16). The presence of an OH group in phenol 5 is sufficient to drive a complete site-selectivity at position 2 (entry 17, Table 8) in the Pd catalyzed reaction with phenylacetylene.
| Entry | Substrate | Coupling reagent | Conditionsa | Position selectivity (%) |
|---|---|---|---|---|
| a (a) Pd-PEPPSI-IPent (2 mol%), THF, 50 °C, 3 h. (b) [PdCl(η3-C3H5)]2 (1 mol%), ferrocenylpolyphosphine 1,2-bis(diphenylphosphino)-1-(diisopropylphosphino)-4-tertbutylferrocene (2 mol%), tBuONa (1.4 equiv.), toluene, 115 °C, 20 h. (c) Mg powder, CuCl, LiCl, 1,3-dimethyl-2-imidazolidinone, 55 °C, 17 h. (d) [Ni(triphos)Cl]PF6(triphos = bis(2-(diphenylphosphino)ethyl)phenylphosphine) (0.5 mol%), Et2O, 0 °C → reflux. (e) Pd(OCOCF3)2 (4 mol%), SPhos (8 mol%), K2CO3 (3 equiv.), 18-crown-6 (5 mol%), scCO2, 110 bar, 110 °C, 24 h. (f) NiCl2(dppbz) (5 mol%), toluene, rt, 22 h. (g) Electrolysis. (h) Co2(CO)8 (7.5 mol%), K2CO3, MeOH, 62 °C, 4 h. (i) hν (350 nm), hexane, 15 h. (j) PdCl2(CH3CN)2 (2 mol%), Cy-DHTP·HBF4 (4 mol%), t-BuOLi toluene, reflux, 45 min. | ||||
| 1 | 1 | PhMgBr | a20a | 2 (0), 3 (70), 2 + 3 (30) |
| 2 | 1 | C6H5NH2 | b20b | 2 (91), 3 (9), 2 + 3 (0) |
| 3 | 1 | 4-MeC6H4NH2 | b20b | 2 (83), 3 (17), 2 + 3 (0) |
| 4 | 1 | Me3SiCl | c20c | 2 (0), 3 (0), 2 + 3 (100) |
| 5 | 1 | EtMgBr | d20d | 2 (5), 3 (95), 2 + 3 (0) |
| 6 | 1 | PhB(OH)2 | e20e | 2 (0), 3 (0), 2 + 3 (100) |
| 7 | 2 | PhMgBr | a20a | 2 (0), 5 (21), 2 + 5 (79) |
| 8 | 3 | PhMgBr | a20a | 3 (0), 4 (12), 3 + 4 (88) |
| 9 | 3 | C6H5NH2 | b20b | 3 (50), 4 (50), 3 + 4 (0) |
| 10 | 3 | 4-MeC6H4NH2 | b20b | 3 (86), 4 (14), 3 + 4 (0) |
| 11 | 3 | Me3SiCl | c20c | 3 (0), 4 (0), 3 + 4 (100) |
| 12 | 4 | PhMgBr | a20a | 2 (0), 4 (30), 2 + 4 (70) |
| 13 | 4 | nC12H25MgBr | f20f | 2 (45), 4 (0), 2 + 4 (55) |
| 14 | 4 | — | g20g | 2 (13), 4 (0), 2 + 4 (87) |
| 15 | 4 | Methyl oxirane + CO (1 atm) | h20h | 2 (100), 4 (0), 2 + 4 (0) |
| 16 | 4 | Cp*Re(CO)3 | i13 | 2 (100), 4 (0), 2 + 4 (0) |
| 17 | 5 |
|
j21 | 2 (100), 3 (0), 2 + 3 (0) |
As apparent from Table 8, the (catalytic) thermal Ar–Cl selective activation in compounds 1–5 is rather complex although scarce data are available for compound 2. The site-selectivity observed in compound 1 depends on the nucleophile used and it is never complete and the reaction is clean only when forming disubstituted products. The single case described for 2 points toward a meta selectivity although 79% of disubstituted product was also present. Again, in derivative 3 the reaction is not clean but meta adducts are preferred with respect to the para derivatives. The best results were found in the case of 4 where the ortho selectivity is exclusive in some cases. Interestingly, the presence of an OH group in place of a methoxy group increases dramatically the selectivity towards the ortho substitution (compare the case of dichlorinated 1 with 5).
The rationalization of the photochemistry observed in compounds 1–5 based on computational analysis is not trivial. In each of the solvents studied two possible triplet states are obtained. It is reasonable to assume that if the energy of one of the triplets is at least lower than 2 kcal mol−1 this should be the reacting excited state and the Ar–Cl cleavage should arise from it. It is interesting to note that calculations suggest a very similar selectivity in the homolytic and in the heterolytic cleavage. This means that in the excited state of compounds 1–5, the Ar–Cl bond cleaved is the same independent of the mechanism operating. This is in fair agreement with what was observed experimentally (see Tables 2–6).
In addition, in the case of compounds having a chloro atom at the ortho position (1–2, 4–5) this halogen is always the preferred leaving group of the reaction. As for compound 3 the cleavage of the chlorine at the para position is slightly preferred with respect to the meta position although the difference between the two most stable structures is very low (<1 kcal mol−1).
The steady-state experiments on compounds 1–5 pointed to a non-selective Ar–Cl cleavage in an almost unpredictable manner. A different site-selectivity was actually found in isomers 1 and 2 where in both cases the chlorine atoms were in the ortho and meta positions with respect to the methoxy group. In fact, in the first case the total absence of selectivity resulted in cyclohexane and in MeOH. In the latter case, however, the ortho cleavage is markedly preferred independent of the medium used. In compound 3, the meta cleavage was indeed preferred upon photohomolysis and exclusively upon photoheterolysis. In the latter case, however, the further dechlorination was too efficient and avoids any synthetic use of the process. The ideal case is that of compound 4 where the complete ortho activation and the limited formation of anisole allowed us to perform a clean site-selective arylation to obtain biphenyl 10.
Quite surprisingly, the selectivity obtained in the case of phenol 5 is very poor (as the photoreactivity) and not better than the corresponding methyl ether 1. This is in contrast to what is usually reported under metal-catalyzed conditions where the OH group usually exerts a marked effect in directing the reactivity toward the ortho position (see also Table 8).21,22–24
5. Conclusion
Although the photoinduced cleavage of the Ar–Cl bond in compounds 1–5 may take place via two different mechanisms the site-selective cleavage of such a bond is a rare occurrence. The versatility offered by the use of different metal-catalysts is indeed superior for the Ar–Cl activation, albeit as apparent from Table 8 a selective reaction is not an easy task. With particular regard to the substrates studied, the high photoreactivity of monochlorinated anisoles (see Table 1) formed after the first chlorine atom detachment is a serious drawback even if the cleavage of the Ar–Cl bond was selective (see the case of compound 4). As a general comment on DFT calculations, it seems apparent that in all cases the homolytic and heterolytic pathways in excited 1–5 were successfully predicted as well as the related energy of the singlet and triplet states of phenyl cations (Fig. 7). As for the last point, the effect of the substituents on the energy of the disubstituted phenyl cations is still an intriguing issue since the (relative) position/nature of such substituents may exert a very peculiar effect (compare the case of 14+ and 15+). The computational method, however, suffers when predicting which Ar–Cl bond cleavage is more feasible since it seems to overestimate in each case the contribution of the Ar–Clortho and Ar–Clpara cleavage; the contribution, however, of competitive photophysical processes may play likewise a crucial role in the observed photochemistry. In summary, the use of direct photocleavage of Ar–X bonds in polyhalogenated derivatives under uncatalyzed metal-free conditions is still in its infancy. A more accurate computational method is required, however, in order to select promising substrates worth to be investigated. An alternative solution could be the use of different kinds of halogens taking into account that the more labile Ar–X bond does not necessarily undergo cleavage in the reaction (see the case of haloanilines where in protic media fluoroanilines were more reactive than the corresponding iodoanilines4).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Davide Ravelli and Dr Stefano Protti (University of Pavia) for fruitful discussion.This work was funded by the CINECA Supercomputer Center, with the computer time granted by ISCRA projects (code: HP10CN4237 and HP10CBEIAU).
Notes and references
- (a) H. Amii and K. Uneyama, C−F Bond Activation in Organic Synthesis, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed Modern Arylation Methods, ed. L. Ackermann, Wiley-VCH Verlag GmbH & Co, 2009, pp. 513–535 Search PubMed (b) Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, Wiley Interscience, New York, 2002, vol. 1 Search PubMed; (c) Top. Curr. Chem., in Cross-Coupling Reactions–A Practical Guide, ed. N. Miyaura, Springer, Heidelberg, 2002, vol. 219 Search PubMed.
- See for example: J. Blum, O. Berlin, D. Milstein, Y. Ben-David, B. C. Wassermann, S. Schutte and H. Schumann, Palladium-Catalyzed Cross-Methylation of Aryl Chlorides by Stabilized Dimethylaluminium and -Gallium Reagents, Synthesis, 2000, 571–575 CrossRef CAS; S. M. Dirk, D. W. Price Jr., S. Chanteau, D. V. Kosynkin and J. M. Tour, N-Acyliminium ion chemistry and palladium catalysis: a useful combination to obtain bicyclic heterocycles, Tetrahedron, 2001, 57, 5109–5121 CrossRef; X. Mei, A. T. August and C. Wolf, Regioselective Copper-Catalyzed Amination of Chlorobenzoic Acids: Synthesis and Solid-State Structures of N-Aryl Anthranilic Acid Derivatives, J. Org. Chem., 2006, 71, 142–149 CrossRef PubMed; T. Schaub, M. Backes and U. Radius, Catalytic C–C Bond Formation Accomplished by Selective C−F Activation of Perfluorinated Arenes, J. Am. Chem. Soc., 2006, 128, 15964–15965 CrossRef PubMed; T. Wang, B. J. Alfonso and J. A. Love, Platinum(II)-Catalyzed Cross-Coupling of Polyfluoroaryl Imines, Org. Lett., 2007, 9, 5629–5631 CrossRef PubMed; L. Ackermann and A. Althammer, Domino N-H/C-H bond activation: palladium-catalyzed synthesis of annulated heterocycles using dichloro(hetero)arenes, Angew. Chem., Int. Ed., 2007, 46, 1627–1629 CrossRef PubMed; I. N. Houpis, J.-P. V. Hoeck and U. Tilstam, Carboxylate-Directed Kumada Coupling of an Acetaldehyde Synthon with 2-Bromobenzoates Used towards the Synthesis of Isochromanes, Synlett, 2007, 2179–2184 CrossRef; Y. Yamamoto and K. Hattori, Synthesis of multiply functionalized benzenes via ruthenium-catalyzed cycloaddition of diiododiynes, Tetrahedron, 2008, 64, 847–855 CrossRef; J. Almond-Thynne, D. C. Blakemore, D. C. Pryde and A. C. Spivey, Site-selective Suzuki-Miyaura coupling of heteroaryl halides - understanding the trends for pharmaceutically important classes, Chem. Sci., 2017, 8, 40–62 RSC.
- S. M. Senaweera, A. Singh and J. D. Weaver, Photocatalytic Hydrodefluorination: Facile Access to Partially Fluorinated Aromatics, J. Am. Chem. Soc., 2014, 136, 3002–3005 CrossRef CAS PubMed.
- M. Freccero, M. Fagnoni and A. Albini, Homolytic vs. Heterolytic Paths in the Photochemistry of Haloanilines, J. Am. Chem. Soc., 2003, 125, 13182–13190 CrossRef CAS PubMed.
- A. Albini and M. Fagnoni, Photochemically-generated intermediates in synthesis, John Wiley & Sons, Hoboken, 2013, pp. 260–301 Search PubMed.
- V. Dichiarante and M. Fagnoni, Aryl Cation Chemistry as an Emerging Versatile Tool for Metal-Free Arylations, Synlett, 2008, 787–800 CAS.
- (a) S. Lazzaroni, D. Ravelli, S. Protti, M. Fagnoni and A. Albini, Photochemical synthesis: Using light to build C-C bonds under mild conditions, C. R. Chim., 2017, 20, 261–271 CrossRef CAS; (b) V. Dichiarante, S. Protti and M. Fagnoni, Phenyl cation: a versatile intermediate, J. Photochem. Photobiol., A, 2017, 339, 103–113 CrossRef CAS.
- S. Protti, M. Fagnoni, M. Mella and A. Albini, Aryl Cations from Aromatic Halides. Photogeneration and Reactivity of 4-Hydroxy(methoxy)phenyl Cation, J. Org. Chem., 2004, 69, 3465–3473 CrossRef CAS PubMed.
- C. Raviola, F. Chiesa, S. Protti, A. Albini and M. Fagnoni, On the Route to the Photogeneration of Heteroaryl Cations. The Case of Halothiophenes, J. Org. Chem., 2016, 81, 6336–6342 CrossRef CAS PubMed.
- E. Fasani, M. Mella, D. Caccia, S. Tassi, M. Fagnoni and A. Albini, The photochemistry of lomefloxacin. An aromatic carbene as the key intermediate in photodecomposition, Chem. Commun., 1997, 1329–1330 RSC.
- M. H. L. Stegeman, W. J. G. M. Peijnenburg and H. Verboom, A Quantitive Structure-Activity relationship for direct photohydrolysis of met-substituted halobenzene in water, Chemosphere, 1993, 26, 837–849 CrossRef CAS.
- M. Mansour, H. Parlar and F. Korte, Photodechlorierungsreaktionen an Dichloranisolen und Dichlortoluolen in Gegenwart von Protonendonatoren, Naturwissenschaften, 1979, 66, 579–580 CrossRef CAS.
- A. Aballay, E. Clot, O. Eisenstein, M.-T. Garland, F. Godoy, A. U. Klahn, J. H. Muñozc and B. Oelckersa, Selectivity in C–Cl bond activation of dichloroarenes by photogenerated Cp*Re(CO)2: combined experimental and DFT studies, New J. Chem., 2005, 29, 226–231 RSC.
- V. Dichiarante, D. Dondi, S. Protti, M. Fagnoni and A. Albini, A meta effect in organic photochemistry? The case of SN1 reactions in methoxyphenyl derivatives, J. Am. Chem. Soc., 2007, 129, 5605–5611 CrossRef CAS PubMed . Correction: 11662.
- A. Kar, N. Mangu, H.-M. Kaise and M.-K. Tse, Gold-catalyzed direct oxidative coupling reactions of non-activated arenes, J. Organomet. Chem., 2009, 694, 524–537 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- C. M. Previtali and T. W. Ebbesen, Laser flash photolysis of chlorobenzene in polar solvents, J. Photochem., 1985, 30, 259–267 CrossRef CAS.
- K.-L. Han and G. Z. He, Photochemistry of aryl halides: Photodissociation dynamics, J. Photochem. Photobiol., C, 2007, 8, 55–66 CrossRef CAS.
- C. Raviola, D. Ravelli, S. Protti, A. Albini and M. Fagnoni, Conditions and Edges for the Photochemical Generation of Short-Lived Aryl Cations: A Computational Approach, Synlett, 2015, 471–478 CAS.
- (a) B. J. Groombridge, S. M. Goldup and I. Larrosa, Selective and general exhaustive cross-coupling of di-chloroarenes with a deficit of nucleophiles mediated by a Pd–NHC complex, Chem. Commun., 2015, 51, 3832–3834 RSC; (b) M. Platon, J. Roger, S. Royer and J.-C. Hierso, Palladium C–N bond formation catalysed by air-stable robust polydentate ferrocenylphosphines: a comparative study for the efficient and selective coupling of aniline derivatives to dichloroarene, Catal. Sci. Technol., 2014, 4, 2072–2080 RSC; (c) T. Kitamura, K. Gondo and T. Katagiri, Synthesis of 1,2-Bis(trimethylsilyl)benzene Derivatives from 1,2-Dichlorobenzenes Using a Hybrid Metal Mg/CuCl in the Presence of LiCl in 1,3-Dimethyl-2-imidazolidinone, J. Org. Chem., 2013, 78, 3421–3424 CrossRef CAS PubMed; (d) G. S. Reddy and W. Tam, Monoalkylation of dichloroarenes with Grignard reagents catalyzed by a nickel complex, Organometallics, 1984, 3, 630–632 CrossRef CAS; (e) I. V. Kuchurov, A. A. Vasil'ev and S. G. Zlotin, The Suzuki–Miyaura cross-coupling of bromo- and chloroarenes with arylboronic acids in supercritical carbon dioxide, Mendeleev Commun., 2010, 20, 140–142 CrossRef CAS; (f) J.-R. Wang and K. Manabe, High Ortho Preference in Ni-Catalyzed Cross-Coupling of Halophenols with Alkyl Grignard Reagents, Org. Lett., 2009, 11, 741–744 CrossRef CAS PubMed; (g) M. Kimura, H. Miyahara, N. Moritani and Y. Sawaki, Electroreductive dehalogenation of chlorinated aromatic ethers. Unexpected electrogenerated base-catalyzed reactions, J. Org. Chem., 1990, 55, 3897–3902 CrossRef CAS; (h) T. S. Khaibulova, I. A. Boyarskaya, E. Larionov and V. P. Boyarskiy, Cobalt-Catalyzed Methoxycarbonylation of Substituted Dichlorobenzenes as an Example of a Facile Radical Anion Nucleophilic Substitution in Chloroarenes, Molecules, 2014, 19, 5876–5897 CrossRef PubMed.
- M. Yamaguchi, T. Akiyama, H. Sasou, H. Katsumata and K. Manabe, One-Pot Synthesis of Substituted Benzo[b]furans and Indoles from Dichlorophenols/Dichloroanilines Using a Palladium–Dihydroxyterphenylphosphine Catalyst, J. Org. Chem., 2016, 81, 5450–5463 CrossRef CAS PubMed.
- K. Kobayashi, M. Arisawa and M. Yamaguchi, GaCl3-Catalyzed Ortho-Ethynylation of Phenols, J. Am. Chem. Soc., 2002, 124, 8528–8529 CrossRef CAS PubMed.
- B. Li, J. Lan, D. Wu and J. You, Rhodium(III)-Catalyzed ortho-Heteroarylation of Phenols through Internal Oxidative C-H Activation: Rapid Screening of Single-Molecular White-Light-Emitting Materials, Angew. Chem., Int. Ed., 2015, 54, 14008–14012 CrossRef CAS PubMed.
- J. Hu, E. A. Adogla, Y. Ju, D. Fan and Q. Wang, Copper-catalyzed ortho-acylation of phenols with aryl aldehydes and its application in one-step preparation of xanthones, Chem. Commun., 2012, 48, 11256–11258 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Photophysical data, computational details and copies of 1H and 13C NMR spectra of the synthesized compounds. See DOI: 10.1039/c7pp00372b |
| This journal is © The Royal Society of Chemistry and Owner Societies 2018 |