
Synthesis and biological assessment of 3,7-dihydroxytropolones†
D. R.
Hirsch
ab,
D. V.
Schiavone
ab,
A. J.
Berkowitz
ab,
L. A.
Morrison
c,
T.
Masaoka
d,
J. A.
Wilson
e,
E.
Lomonosova
c,
H.
Zhao
f,
B. S.
Patel
c,
S. H.
Datla
c,
S. G.
Hoft
c,
S. J.
Majidi
c,
R. K.
Pal
ag,
E.
Gallicchio
abg,
L.
Tang
f,
J. E.
Tavis
c,
S. F. J.
Le Grice
d,
J. A.
Beutler
 e and
R. P.
Murelli
*ab
e and
R. P.
Murelli
*ab
aDepartment of Chemistry, Brooklyn College, The City University of New York, Brooklyn, New York, 11210, USA. E-mail: rpmurelli@brooklyn.cuny.edu
bPhD Program in Chemistry, The Graduate Center of The City University of New York, New York, New York 10016, USA
cDepartment of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, St. Louis, Missouri 63104, USA
dBasic Research Laboratory, Center for Cancer Research, National Cancer Institute, Frederick, MD 21702-1201, USA
eMolecular Targets Laboratory, Center for Cancer Research, National Cancer Institute, Frederick, MD 21702-1201, USA
fDepartment of Molecular Biosciences, The University of Kansas, Lawrence, Kansas 66045, USA
gPhD Program in Biochemistry, The Graduate Center of The City University of New York, New York, New York 10016, USA
First published on 24th October 2017
Abstract
3,7-Dihydroxytropolones (3,7-dHTs) are highly oxygenated troponoids that have been identified as lead compounds for several human diseases. To date, structure–function studies on these molecules have been limited due to a scarcity of synthetic methods for their preparation. New synthetic strategies towards structurally novel 3,7-dHTs would be valuable in further studying their therapeutic potential. Here we describe the successful adaptation of a [5 + 2] oxidopyrilium cycloaddition/ring-opening for 3,7-dHT synthesis, which we apply in the synthesis of a plausible biosynthetic intermediate to the natural products puberulic and puberulonic acid. We have also tested these new compounds in several biological assays related to human immunodeficiency virus (HIV), hepatitis B virus (HBV) and herpes simplex virus (HSV) in order to gain insight into structure-functional analysis related to antiviral troponoid development.
Introduction
7-Hydroxytropolones (αHTs) and 3,7-dihydroxytropolones (3,7-dHTs) (Scheme 1A) are troponoids with three and four contiguous oxygen atoms, respectively, and display an extraordinarily broad range of biological activity.1 While this activity is most often attributed in both cases to their ability to serve as metal-binding fragments for many physiologically relevant dinuclear metalloenzymes (Scheme 1A), stark differences in bioactivity have been observed between them. For example, αHTs appear to be substantially more potent than 3,7-dHTs as inhibitors of HIV ribonuclease H,2,3 a promising target for HIV antivirals that remains untargeted clinically, as well as aminoglycoside-2′′-O-nucleotidyltransferase,4 one of the most common determinants of enzyme-dependent aminoglycoside resistance in Pseudomonas aeruginosa. On the other hand, the 3,7-dHT natural product puberulic acid has demonstrated potent antimalarial activity, with selectivity on par with clinical agent artesunate, whereas in similar assays αHTs have substantially lower potency and selectivity.5 3,7-dHT also possesses activity against B16 melanoma cells an order of magnitude greater than αHTs, and increases the lifespan of mice bearing B16 melanoma comparably to mice administered the chemotherapeutic agent mitomycin C.6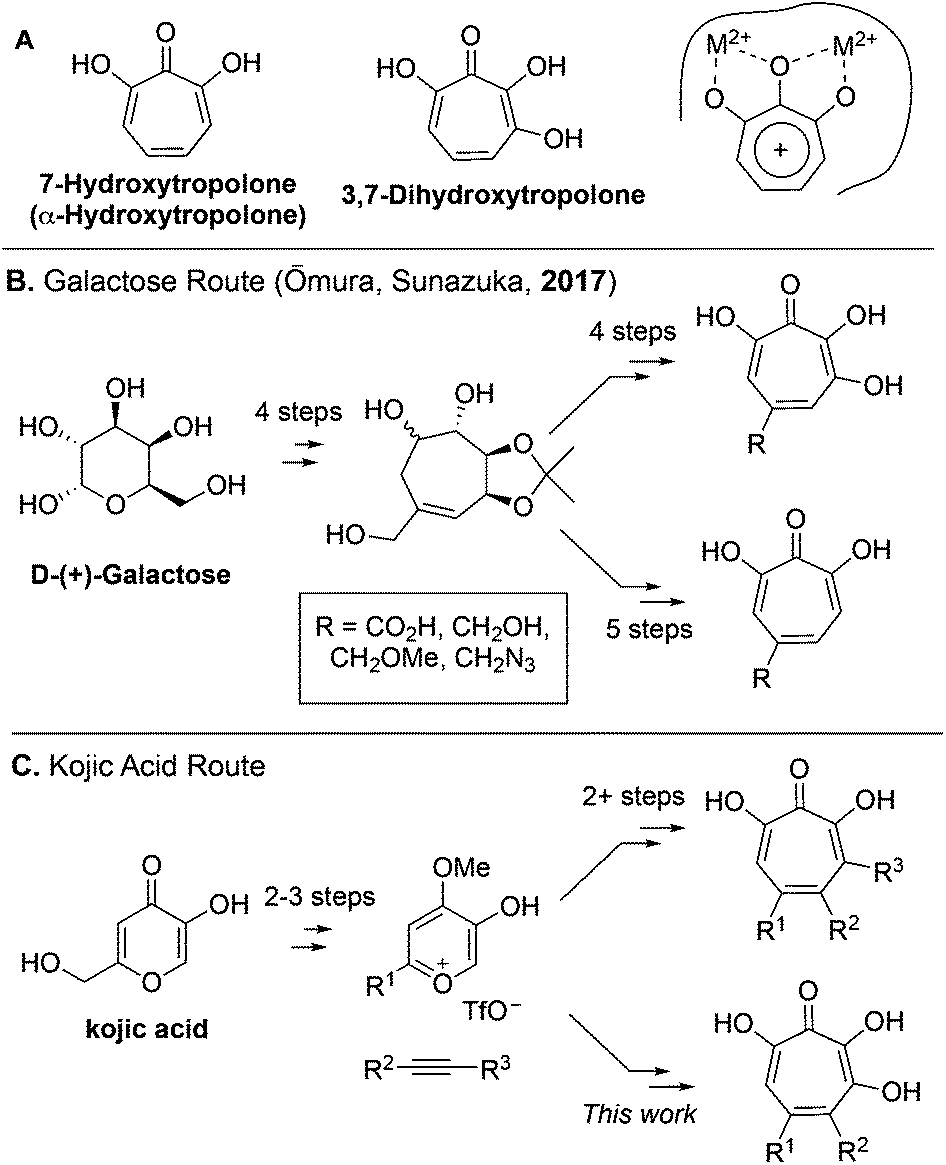 | ||
| Scheme 1 (A) Contiguously hydroxylated tropolones and common mode of metalloenzyme binding. (B) A divergent, unifying galactose approach towards both tropolone classes. (C) Overview of kojic acid route developed by our lab and extended in this study to 3,7-dHTs. | ||
In order to better understand the nuances of structure-bioactivity differences between these classes of troponoids, efficient synthetic methods are needed to access both classes, and of particular value would be those that also allow comparable substitution.7 During the preparation of this manuscript, a galactose-based method previously used by Sunazuka and Omura to synthesize puberulic acid8 was further adapted to develop a small library of both 3,7-dHTs and αHTs (Scheme 1B), as well as other related troponoids.9 Testing of these molecules for their antimalarial activity revealed that a carboxylic acid was key to providing selectivity against Plasmodium falciparum K1 versus MRC-5 cells, and it did so whether on an αHT or a 3,7-dHT, but provided more selectivity with 3,7-dHT. These studies highlight the value in a divergent, unifying strategy in SAR determination of oxygenated troponoids, and their importance in assessing the inhibitory potential of these oxygenated troponoids.
Our lab has previously reported an extremely efficient oxidopyrylium cycloaddition/ring-opening strategy for αHT synthesis that has to date generated over 50 published αHTs with varying substitution patterns,10,11 and has been used to provide SAR and pursue synthetic chemistry-based optimization studies related to various human diseases.12–18 We herein report the adaptation of this route to access a series of 3,7-dHTs. We highlight this strategy through the synthesis of a potential biosynthetic precursor to the 3,7-dHT natural products puberulonic and puberulic acid. The molecules were assessed as part of biochemical and biological studies related to HIV, hepatitis B virus (HBV), and herpes simplex virus (HSV)-1 and -2. These studies help provide an understanding of the advantages and disadvantages of 3,7-dHTs and αHTs as chemotypes for drug discovery and development.
Results and discussion
Synthesis
Our synthesis started with a cycloaddition between oxidopyrylium triflate salt 1a, which can be made on gram scale in 3 steps from commercially-available kojic acid,19 and iodopropiolates (Scheme 2A, 2a/b), which are prepared from the corresponding propiolates using silver nitrate and N-iodosuccinimide.20,21 Bromopropiolates are also capable of efficient oxidopyrylium cycloaddition and have been used in the synthesis of bromohydroxytropolones,14 but their volatility and strong lachrymator properties pose technical challenges. The iodobicycles can then be converted into methoxybicycles through a DMAP-catalyzed methanolysis (3a/b → 4a/b). These conditions were deemed necessary when we found that Brønsted base-mediated methanolysis led to dimethyl acetals (e.g.9), while DMAP incorporation was observed as the major product when chloroform was employed as the solvent in an attempt at hydrolysis (8, Scheme 2B). Interestingly, subsequent ring-opening attempts on these bicycles (4a/b → 5a/b) using sulfonic acids led to formation of oxidopyrylium dimers as the major product, as observed by crude 1H NMR (e.g.7a, Scheme 2C). We hypothesize that this may proceed via protonation and elimination of the β-methoxyenoate, which would lead to a rapidly dimerizing oxidopyrylium ylide.22 Fortunately, an alternative procedure using the Lewis acid boron trichloride promoted clean conversion to 3,7-dimethoxytropolones 5a and 5b, which were subsequently converted to the 3,7-dHTs 6a and 6b using hydrobromic acid in acetic acid at elevated temperatures. Additionally, in the presence of excess water along with extended reaction times, methyl ester 5a was readily decarboxylated (6c, Scheme 2D), which can be attributed to the participation of a β-keto acid tautomer. Thus, while the ester provides a convenient synthetic handle for efficient oxidopyrylium cycloaddition and subsequent iodide for methoxide exchange, it can be readily removed if desired. | ||
| Scheme 2 (a) Formation of 3,7-dHTs via oxidopyrylium salt 1a and iodoalkynes 2a and 2b. (b) Results from initial attempts at methanol-iodide exchange. (c) Brønsted acid-mediated cycloreversion of oxabicyclic intermediates 3. (d) Acid-mediated hydrolysis and decarboxylation of 5a. Reagents and conditions: (a) i-Pr2NPh, 2a/b, CH2Cl2, 120 °C, 20 min (3a = 57%, 3b = 95%). (b) MeOH/DMAP, 120 °C, 15 min (4a = 94%, 4b = 73%). (c) BCl3, CH2Cl2, r.t., 10 min, (5a = 97%, 5b = 81%). (d) HBr/AcOH, 120 °C, 45 min (6a = 55%, 6b = 87%). (e) DMAP/H2O, CDCl3, 100 °C, 90 min, 16%. (f) K2CO3/MeOH, r.t., 67%. (g) HBr/AcOH/H2O, 120 °C, 60 min, 45%. | ||
With an efficient method for 3,7-dHT synthesis in hand, we sought to test the new strategy in total synthesis. The two most widely studied 3,7-dHTs are puberulonic and puberulic acid, which have been isolated from the fungus Penicillum puberulum.23 These natural products are also close structural homologues of the 6-hydroxytropolones stipitatonic and stipitatic acid,24 the focus of recent biosynthetic studies by Cox and coworkers.25 These studies demonstrated that stipitatic acid is formed from stipitatonic acid, which is, in turn, formed from the lactone stipitalide (Scheme 3). While a similar biosynthetic pathway seems plausible for puberulonic and puberulic acid, the 3,7-dHT stipitalide homologue 6d is unknown. Access to this molecule could be useful in elucidating the biosynthesis of puberulic and puberulonic acid, and our synthetic strategy appeared uniquely well suited for this task. Thus we set out to conduct a total synthesis of 6d.
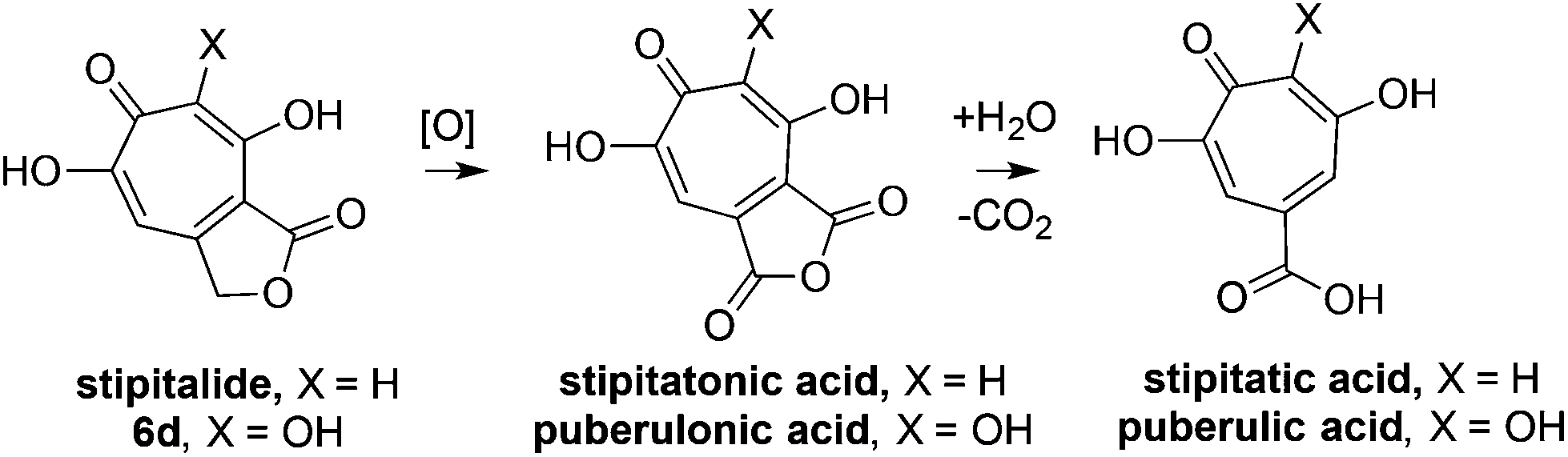 | ||
| Scheme 3 Biogenesis of 6-hydroxytropolones stipitatonic acid and stipitatic acid from stipitalide and related 3,7-dHTs. | ||
We began our synthesis with the chloromethylene-containing oxidopyrylium salt 1b (Scheme 4). Initial attempts towards bicycle 3c using our prior conditions (‘a’, Scheme 4) lead to a significant amount of compound 3b, which created purification challenges given the similar polarities of 3b and 3c. We hypothesized that this product was formed through a hydride transfer from the N,N-diisopropylaniline (Scheme 2A),26 and thus, leveraging the readily reversible nature of this class of oxidopyrylium ylides (formed following deprotonation of 1b),10 we synthesized and isolated dimer 7b as the source of the ylide.27 Indeed, this alternative procedure led to the dihalogen 3c without any noticeable 3b. While this intermediate offered two electrophilic handles, namely a β-iodoacrylate for methanolysis and a primary alkyl chloride for eventual lactonization, we reasoned that the alkyl chloride would be stable to the methanolysis conditions because of the adjacent tertiary centre. Indeed, methanolysis of 3c proceeded efficiently while maintaining the chloride's integrity, even though higher temperatures were needed for this transformation than for 3a/b. Subsequent ring-opening afforded methoxytropolone 5c, which was then advanced to 6d through a three-step acetolysis/lactonization/demethylation sequence. During the course of these studies, we also discovered that reverse-phase chromatography of 5c facilitated on-column lactonization to afford 5e, providing a more direct synthetic method.
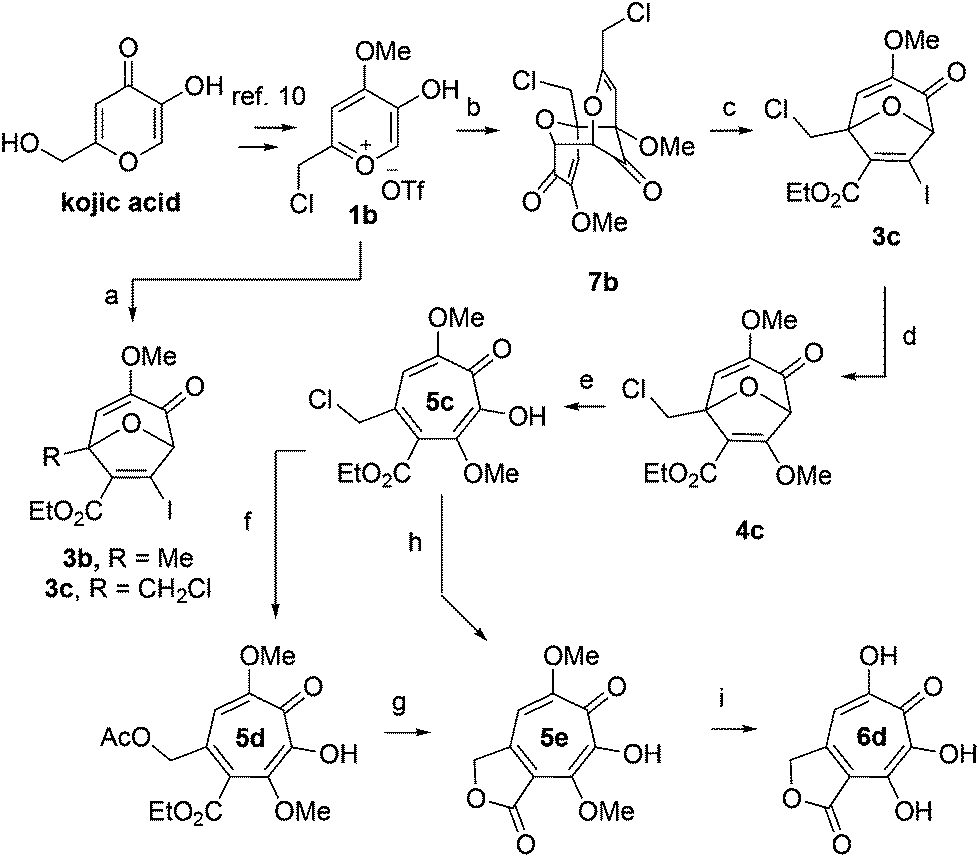 | ||
Scheme 4 Synthesis of 3,7-dHT 6d. Reagents and conditions: (a) iPr2NPh, 2b, CH2Cl2, 100 °C, 1 h, 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 3c:3b observed by crude 1H NMR. (b) NEt3, CH2Cl2, 85%. (c) 2b, CH2Cl2, 100 °C, 1 h, 71%. (d) DMAP, MeOH, 120 °C, 5 min, 61%. (e) BCl3, CH2Cl2, 0 °C, 5 min, 89%. (f) NaOAc, AcOH, r.t., 15 h, 93%. (g) NaOH (aq.), r.t., 3 h, 91%. (h) C18 silica, MeCN/H2O/TFA, 81%. (i) HBr/AcOH, 120 °C, 45 min, 35%. :1 ratio of 3c:3b observed by crude 1H NMR. (b) NEt3, CH2Cl2, 85%. (c) 2b, CH2Cl2, 100 °C, 1 h, 71%. (d) DMAP, MeOH, 120 °C, 5 min, 61%. (e) BCl3, CH2Cl2, 0 °C, 5 min, 89%. (f) NaOAc, AcOH, r.t., 15 h, 93%. (g) NaOH (aq.), r.t., 3 h, 91%. (h) C18 silica, MeCN/H2O/TFA, 81%. (i) HBr/AcOH, 120 °C, 45 min, 35%. | ||
Biology
One of the more active areas of research in our group has been αHT antiviral development, as several viral nucleases have been identified as targets for these molecules.2,3,13–18 The most widely studied in these contexts is the ribonuclease H function of HIV reverse transcriptase (HIV RT RNaseH),2,3,13,28–30 a promising candidate for therapeutic development that remains untargeted clinically. αHTs are potent inhibitors of the enzyme with 50% inhibition concentrations (IC50) less then 200 nM, and several synthetic αHTs have been developed with modest antiviral activity in cell-based assays.2,13,29 While both 3,7-dHTs and αHTs are known inhibitors of the enzyme, and the collection of literature sources indicate that αHTs are superior inhibitors, head-to-head comparisons of both classes of molecules have not to the best of our knowledge been conducted. We thus tested 3,7-dHTs 6a–6d, αHT congener 10, and methoxytropolone congers 5a and 11 for their ability to inhibit HIV-1 RT RNaseH. Consistent with literature trends, αHT 10 was the most potent of the compounds tested, with a 4–5 fold increase over the 3,7-dHTs (Fig. 1). Methoxytropolones were even less potent, consistent with tropolone activity. Furthermore, while promising cell-based HIV antiviral activity of 3,7-dHT has been described in the literature,3 our synthetic 3,7-dHTs showed no protective effects, possibly due to a combination of weaker anti-RNaseH activity and higher cytotoxicity. Compound 10, which is known to have modest HIV protective effects,13 was tested simultaneously as a positive control. Thus, early indications suggest that the extra hydroxyl in 3,7-dHTs is unfavourable for HIV therapeutic development targeting RNaseH.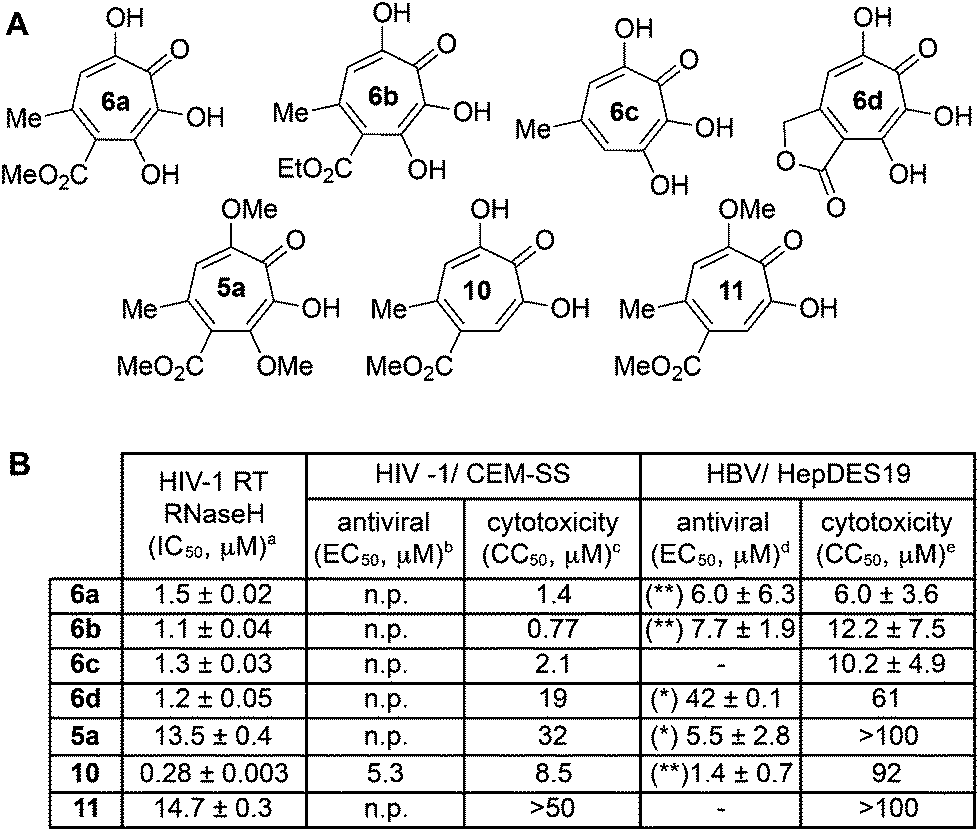 | ||
| Fig. 1 Viral ribonuclease H-related studies. (A) Compounds tested in antiviral assays. (B) Summary of data from RNase H-related biological studies for HIV and HBV. aHIV-1 RT RNaseH IC50 values are reported as the average from triplicate assays ± standard deviation. bConcentration of 50% viral replication suppression (EC50) are reported from a single run. n.p. Reflects not protective. cCC50 represents concentration at 50% cytotoxicity against the host CEM-SS cells, and are calculated from a single experiment. dHBV replication inhibition at 20 μM, unless otherwise defined. ** Indicates that the amount of (+)-strand DNA is <25% and (−)-strand DNA is >60% relative to DMSO control. * Indicates that the amount of (+)-strand DNA is <50% of the amount of (−)-strand DNA relative to DMSO control. – refers to no evidence of selective inhibition of (+)-strand DNA. EC50 values are shown for active compounds, and are reported as the average of two or three dose response curves ± standard deviation. eHepDES19 cytotoxicity values are likewise reported as an average of 2–4 assays ± standard deviation. | ||
Hepatitis B also has an RNase H function that has become a target for antiviral drug development, and αHTs have emerged as one of the most promising scaffolds.31 A 3,7-dHT has never been tested for this activity, and thus we assessed our new compounds’ HBV RNase H-specific antiviral activity by monitoring the amount of viral (+)-DNA strand versus (−)-DNA, since inhibiting RNase H activity suppresses production of the former. As expected, αHT 10 showed only minor cytotoxicity against the host cell line, HepDES19, while selectively inhibiting synthesis of the viral (+)-DNA. Selective inhibition of viral (+)-DNA was also observed with 3,7-dHTs 6a and 6b, although they were significantly more cytotoxic. It is worth noting the structural similarities of the two selective inhibitors, 6a and 6b, to compound 10 and the lack of activity of compound 6c, which could indicate an important role of the carbonyl appendage. Unfortunately, the EC50 values of the viral suppression of 6a/b coupled with their CC50 values revealed virtually no therapeutic window, as compared to the 60-fold window of αHT 10. Thus, while these experiments demonstrate antiviral potential for 3,7-dHTs that may warrant further studies, early indications suggest they are not advantageous over αHTs. On the other hand, the 3,7-dimethoxytropolone 5a displayed moderate inhibition selectivity for (+)-DNA, had an antiviral EC50 value of 5.5 μM, and was non-toxic at concentrations of up to 100 μM. Thus, 3,7-dimethoxytropolone appears to be a viable chemotype for further anti-HBV development.
While 3,7-dHTs had never been tested for anti-HSV activity prior to our studies, αHTs were identified as anti-HSV agents, with the natural product manicol displaying antiviral EC50 values against HSV-1 and -2 of 350 nM and 580 nM, respectively.32 Furthermore, tests against 20 synthetic αHTs synthesized through our oxidopyrylium cycloaddition/ring-opening procedure identified a synthetic αHT with antiviral EC50 values against HSV-1 and -2 of 120 nM and 80 nM, respectively (see 12, Fig. 3).15 In all cases, cytotoxicity was not observed against Vero cells within the 24 hours experiment. We thus tested our library of tropolones and found that 3,7-dHTs 6a, 6b, and 6c all strongly inhibited replication of HSV-1 at a concentration of 5 μM, with suppression levels comparable to the therapeutic anti-HSV agent acyclovir (ACV) and exceeding that of cidofovir (CDV) (Fig. 2A). αHT 10 only showed moderate replication inhibition at 5 μM, and no activity at 1 μM, whereas the activity for 6a–6c was maintained. Methoxytropolones 5a and 11 were the least active compounds among the series tested. 3,7-dHT 6d was inactive, illustrating how changes in the side-chains can impact activity. 3,7-dHTs 6a–6c inhibited HSV-2 replication with comparable suppression levels and potencies, and this activity was maintained against three other HSV-2 clinical isolates at 5 μM (Fig. 2B). The EC50 values of compounds 6a and 6c were comparable to ACV for HSV-1, and showed no cytotoxicity versus the Vero cell line at up to 100 μM for 24 hours (Fig. 2C). Furthermore, while ACV was nearly an order of magnitude less potent against HSV-2 than HSV-1, 6a and 6c maintained significant potency.
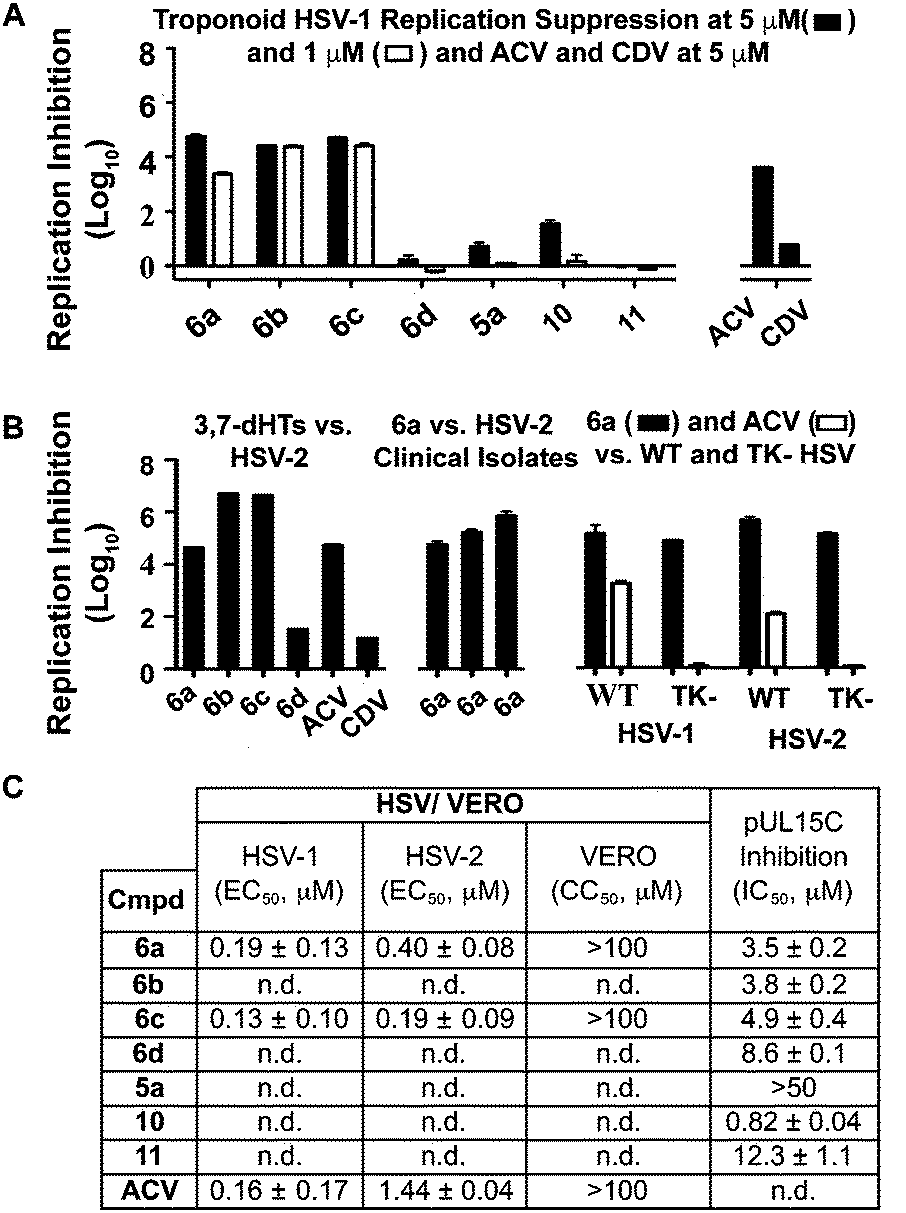 | ||
| Fig. 2 HSV-related studies. (A) HSV-1 suppression by synthetic tropolones at concentrations of 5 and 1 μM. Also shown are clinical anti-HSV agents acyclovir (ACV) and cidofovir (CDV) at 5 μM. (B) Replication inhibition studies of 3,7-dHTs at 5 μM. L/R 6a–d against HSV-2, compound 6a against 3 HSV-2 clinical isolates, and comparisons of 6a and ACV against wild-type (WT) and thymidine kinase negative (TK−) HSV strains resistant to ACV. (C) EC50 values obtained for 6a and 6c against HSV-1 and HSV-2, with data representing the average from 2 dose response curves +/− standard deviation. Also shown are values for ACV obtained previously by Tavis et al. under identical conditions.32 CC50 represents concentration at 50% cytotoxicity against the host Vero cells. n.d. = not determined. | ||
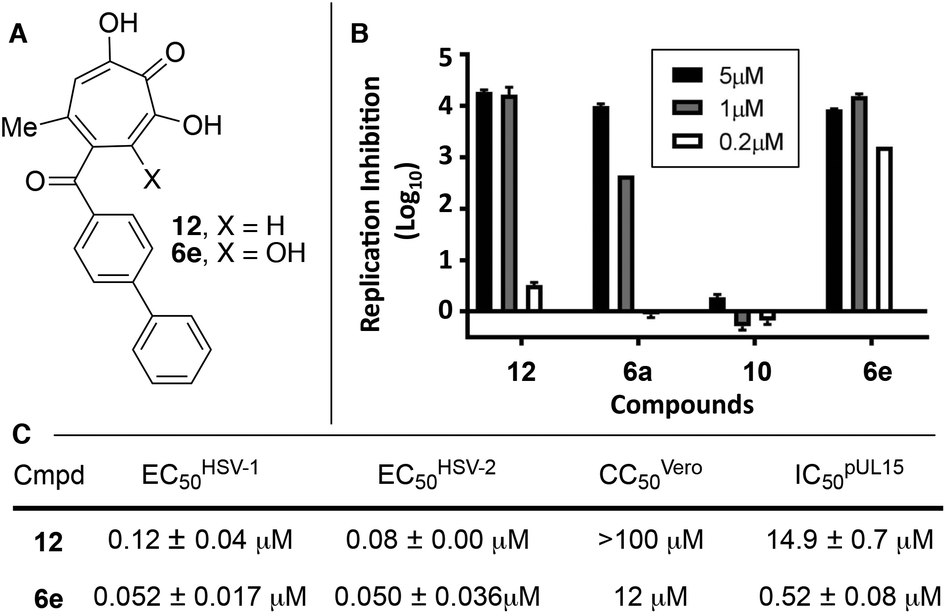 | ||
| Fig. 3 HSV-associated assays of compound 12 and related structures. (A) Structures of most potent anti-HSV molecules. (B) Head-to-head comparison of troponoids as HSV-1 viral replication inhibitors at three concentrations. (C) Potency values for 6e and 12. Values of 12 were reported previously.15 | ||
Although the mechanism of action is currently under investigation, experiments with 6a against the ACV-resistant TK− HSV-1 and HSV-2 strains confirmed a different mechanism of action than ACV (Fig. 2B). One hypothesis is that the molecules could engage one or more viral nucleotidyltransferases.33 For example, the HSV-1 DNA packing terminase pUL15 encodes a C-terminal nuclease activity that can be inhibited by αHTs.14,34 Thus, we tested 3,7-dHTs against recombinant pUL15C and found them to be inhibitors of the enzyme, but with less potency than αHT 10 (Fig. 2C).
Given the SAR suggesting benefits of the additional oxygen for HSV inhibition potency, a 3,7-dHT analogue of the most potent anti-HSV αHT described to date, compound 12, was synthesized (6e, Scheme 5). Anti-HSV-1 activity was assessed in parallel with 3,7-dHT 6a and αHTs 12 and 10 (Fig. 3B). As expected, compound 10 showed no antiviral activity, even at 5 μM, whereas some viral suppression was observed for 12, 6a and 6e. At 1 μM, 6e and 12 antiviral activity remained at maximum, but activity of 6a began to drop. At 200 nM, 6a was inactive, and both 12 and 6e began to lose some activity. EC50 values of 6e were ∼50 nM against both HSV-1 and -2, which is the most potent anti-HSV activity of a tropolone we have found to date. Unfortunately, moderate cytotoxicity of 6e was also observed.
 | ||
| Scheme 5 Synthesis of 3,7-dHT 6e. Reagents and conditions: (a) AgNO3, N-iodosuccinimide, acetone, 0 °C, 4 h, 57%. (b) 2d and 7a, CH2Cl2, 120 °C, 30 min (c) MeOH/DMAP, 70 °C, 15 min, 68% over two steps. (d) BCl3, CH2Cl2, 0 °C, 6 min, 64%. (e) HBr/AcOH, 120 °C, 70 min, 70%. | ||
While increases in potency against HSV were anticipated through additive effects, surprising effects were observed with pUL15 inhibition. While both a change from an αHT to a 3,7-dHT (10 → 6a), and from a methyl ester to a biphenyl ketone (10 → 12) resulted in a decrease in pUL15 potency, changing both provided an increase in potency (10 → 6e). One hypothesis for this change is that the added hydroxyl permits the biphenyl side chain to adopt a new configuration that would promote new favourable contacts (Fig. 4A vs. B). In silico modelling was thus carried out by superimposing the active site of crystal structure of pUL15C (PDB id 4IOX)34 to the active site of the crystal structure of HIV RT RNase H bound to manganese cations and β-thujaplicinol (PDB id 3 K2P).35 The resulting structure of pUL15C was refined by energy minimization and simulated annealing using the Impact molecular modeling program.36 The receptor grid was generated using the default parameters available in the Schrodinger Suite 2016-3 with one special adjustment where metal constraints were applied to allow metal–ligand interaction at the binding site. Glide docking37 were performed with the ligands αHT 12 and 3,7 dHT 6e. Several binding poses were obtained for the molecule 6e, out of which the best binding pose is shown in Fig. 4.
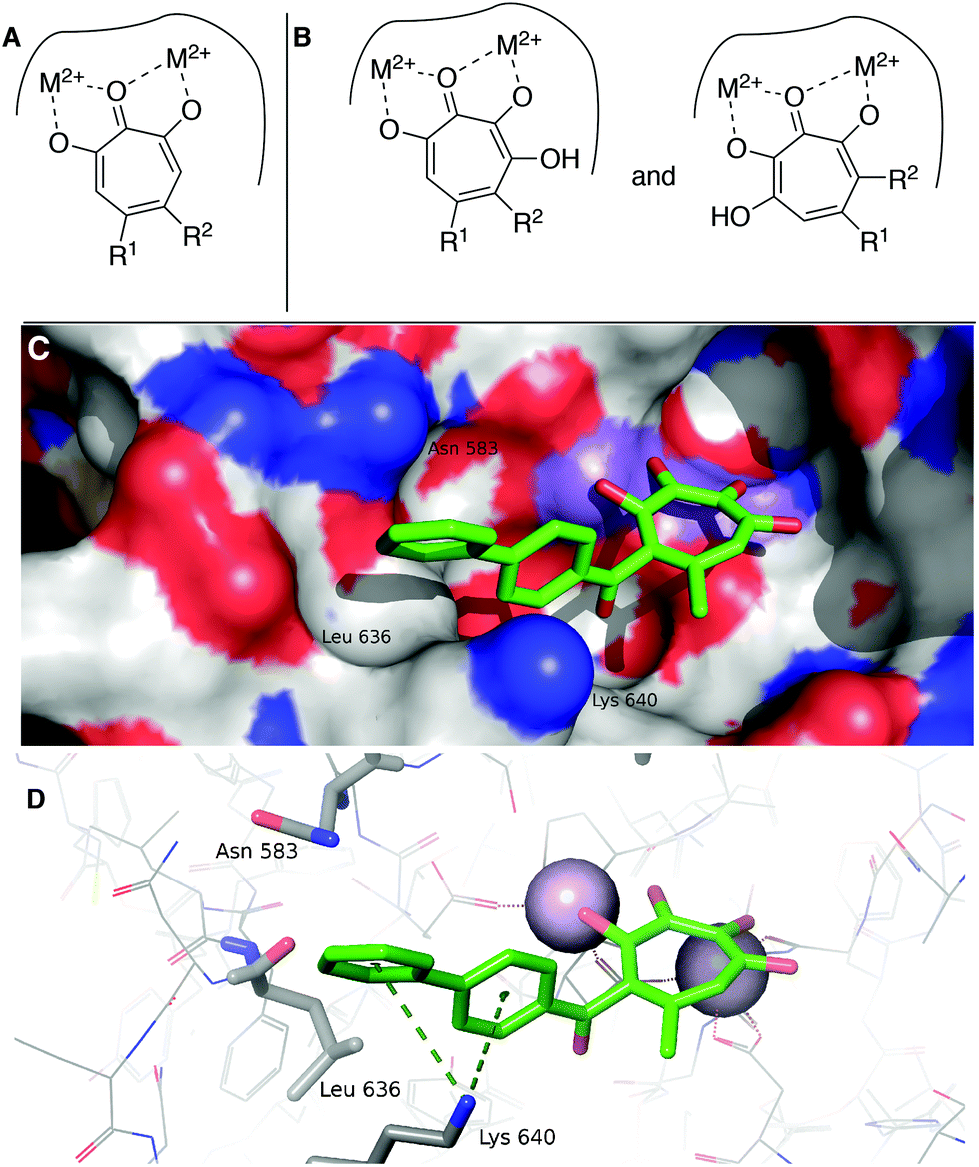 | ||
| Fig. 4 Hypothesized structural basis for potency increases of 6e against pUL15. (A) Single orientation of αHT as opposed to (B) two orientations of 3,7-dHTs. (C,D) The structure of the complex between UL15 (PDB id 4IOX) and 3,7-dHT 6e (green) predicted by molecular docking, showing (C) the biphenyl group of the ligand nested into a hydrophobic groove formed by Lys 640, Asn 583 and Leu 636 and (D) possible π–cation contacts with Lys 640 and close proximity to Asn 583, which could participate in π–NH interactions. | ||
This conformation reveals favorable π–cation interaction between the quaternary ammonium cation of Lys 640 and the phenyl ring π system of the biphenyl group of the ligand. The biphenyl group is thought to be further stabilized by accommodating within a hydrophobic groove formed between Lys 640 and Asn 583 with possible hydrophobic interaction with the Leu 636 present inside the groove. Further stabilization could also be achieved through interactions of the biaryl side chain with Asn 583 through NH–π interactions. These interactions are never observed in in silico modelling with 12. While further studies are needed to understand the specific mechanism of this increase in activity, these analyses highlight the consequences of the additional binding modes allowed by 3,7-dHTs, which might also result in greater likelihood of off-target effects and be a reason for the higher cytotoxicity observed with this class of tropolones.
Conclusions
We have developed an oxidopyrylium cycloaddition/ring-opening approach for the synthesis of 3,7-dHTs. This strategy was applied in the synthesis of a prospective biosynthetic precursor to the natural products puberulonic and puberulic acid. Finally, the new 3,7-dHTs were tested for antiviral activity against HIV, HBV and HSV. Through these studies we found that 3,7-dHTs are weaker inhibitors than αHTs against HIV RNaseH, revealed 3,7-dimethoxytropolone as a promising chemotype for HBV-based antiviral development, and demonstrated that 3,7-dHTs can have greater potency against HSV than analogous αHTs. Furthermore, enzymatic inhibition studies coupled with preliminary in silico modelling highlight how additional configurations of 3,7-dHTs may manifest themselves in unexpected potency increases, and could also be responsible for observed cytotoxicity increases.Author contributions
DH, AB, DS, and RM conducted the work associated with the synthesis. TM, JW, JB, SL obtained data associated with HIV. EL and JT obtained data associated with HBV. LM, BP, HD, SH, and SM obtained data associated with HSV. TM, HZ, LT, and SL obtained data associated with pUL15C. RP and EG are responsible for in silico modelling. DH and RM wrote manuscript with primary assistance and feedback from SL, JB, LM, JT, LT and EG.Conflicts of interest
RM, LM, and JT are coinventors on a patent application that covers anti-HBV and anti-HSV activity of hydroxytropolones.Acknowledgements
DH, AB, DS, RP and RM are grateful for funding from the National Institutes of Health (SC1GM111158), as are EL and JT (R01AI122669) and HZ and LT (R01GM090010). SL, TM, JW and JB acknowledge support from the Intramural Research Program of the National Cancer Institute, National Institutes of Health, Department of Health and Human Services. LM, BP, SH, DG, SH, GH, and SM are funded through the Saint Louis University President's Research Fund and a gift from the Pershing Trust. We also thank Kaelin Bernier, Jil A. W. Daw and Nathan L. Ponzar for technical assistance, and Prof. Tom Kurtzman at Lehman College (CUNY) for computational support. Finally, NMR data of 6d was in part collected at the City University of New York Advanced Science Research Center (CUNY ASRC) Biomolecular NMR facility, and we thank Dr James Aramini for his assistance in the collection of this data.Notes and references
- For a review, see: C. Meck, M. P. D'Erasmo, D. R. Hirsch and R. P. Murelli, Med. Chem. Commun., 2014, 5, 842–852 RSC
.
- J. Didierjean, C. Isel, F. Querré, J. F. Mouscadet, A. M. Aubertin, J. V. Valnot, S. R. Piettre and R. Marquet, Antimicrob. Agents Chemother., 2005, 49, 4884–4894 CrossRef CAS PubMed
- S. R. Budihas, I. Gorshkova, S. Gaidamakov, A. Wamiru, M. K. Bona, M. M. A. Parniak, R. J. Crouch, J. B. McMahon, J. A. Beutler and S. F. J. Le Grice, Nucleic Acids Res., 2005, 33, 1249–1256 CrossRef CAS PubMed
- N. E. Allen, W. E. Alborn Jr., J. N. Hobbs Jr. and H. A. Kirst, Antimicrob. Agents Chemother., 1982, 22, 824 CrossRef CAS PubMed
- M. Iwatsuki, S. Takada, M. Mori, A. Ishiyama, M. Namatame, K. Otoguro, A. Nishihara-Tsukashima, K. Nonaka, R. Masuma, O. Otoguro, K. Shiomi and S. Omura, J. Antibiot., 2011, 64, 183–188 CrossRef CAS PubMed
- K. Sugawara, M. Ohbayashi, K. Shimizu, M. Hatori, H. Kamei, M. Konishi, T. Oki and H. Kawaguchi, J. Antibiot., 2011, 64, 862–868 Search PubMed
- For examples of αHT synthesis, see.
(a) M. G. Banwell, M. P. Collis, G. T. Crisp, J. T. Lambert, M. E. Reum and J. A. Scoble, J. Chem. Soc., Chem. Commun., 1989, 10, 616–617 RSC
- G. Sennari, T. Hirose, M. Iwatsuki, S. Omura and T. Sunazuka, Chem. Commun., 2014, 50, 8715–8718 RSC
- G. Sennari, R. Saito, T. Hirose, M. Iwatsuki, A. Ishayama, R. Hokari, K. Otoguro, S. Omura and T. Sunazuka, Sci. Rep., 2017, 7, 7259 CrossRef PubMed
- C. Meck, N. Mohd and R. P. Murelli, Org. Lett., 2012, 14, 5988–5991 CrossRef CAS PubMed
- Y. D. Williams, C. Meck, N. Mohd and R. P. Murelli, J. Org. Chem., 2013, 78, 11707–11713 CrossRef CAS PubMed
- D. R. Hirsch, G. C. Cox, M. P. D'Erasmo, T. Shakya, T. C. Meck, N. Mohd, G. D. Wright and R. P. Murelli, Bioorg. Med. Chem. Lett., 2014, 24, 4943–4947 CrossRef CAS PubMed
- R. P. Murelli, M. P. D'Erasmo, D. R. Hirsch, C. Meck, T. Masaoka, J. A. Wilson, B. Zhang, R. K. Pal, E. Gallicchio, J. A. Beutler and S. F. J. Le Grice, Med. Chem. Commun., 2016, 9, 1783–1788 RSC
- T. Masaoka, H. Zhao, D. R. Hirsch, M. P. D'Erasmo, C. Meck, B. Varnado, A. Gupta, M. J. Meyers, J. Baines, J. A. Beutler, R. P. Murelli, L. Tang and S. F. J. Le Grice, Biochemistry, 2016, 55, 809–819 CrossRef CAS PubMed
- P. J. Ireland, J. E. Tavis, M. P. D'Erasamo, D. R. Hirsch, R. P. Murelli, M. M. Cadiz, B. S. Patel, A. K. Gupta, T. C. Edwards, M. Korom, E. A. Moran and L. A. Morrison, Antimicrob. Agents Chemother., 2016, 60, 2140–2149 CrossRef CAS PubMed
- G. Lu, E. Lomonosova, X. Cheng, E. A. Moran, M. J. Meyers, S. F. J. Le Grice, C. J. Thomas, J.-K. Jiang, C. Meck, D. R. Hirsch, M. P. D'Erasmo, D. M. Suyabatmaz, R. P. Murelli and J. E. Tavis, Antimicrob. Agents Chemother., 2015, 59, 1070–1079 CrossRef PubMed
- M. J. Donlin, A. Zunica, A. Lipnicky, A. K. Garimallaprabhakaran, A. J. Berkowitz, A. Grigoryan, M. J. Meyers, J. E. Tavis and R. P. Murelli, Antimicrob. Agents Chemother., 2017, 61, e02574–e02516 CrossRef CAS PubMed
- E. Lomonosova, J. Daw, A. K. Garimallaprabhakaran, N. A. Agyemang, Y. Ashani, R. P. Murelli and J. E. Tavis, Antiviral Res., 2017, 144, 164–172 CrossRef CAS PubMed
- P. A. Wender and J. L. Mascarenás, Tetrahedron Lett., 1992, 33, 2115–2118 CrossRef CAS
- U. Halbes-Letinois, J.-M. Weibei and P. Pale, Chem. Soc. Rev., 2007, 36, 759–769 RSC
- P.-C. Qian, R.-J. Song, H.-Y. Liao, H.-T. Wang, Y.-X. Xie, J.-N. Xiang and J.-H. Li, Synthesis, 2015, 3309–3314 CAS
- H.-Y. Lee, H. Y. Kim, B. G. Kim and J. M. Kee, Synthesis, 2007, 2360–2364 CrossRef CAS
-
(a) J. H. Birkinshaw and H. Raistrick, Biochem. J., 1932, 26, 441–453 CrossRef CAS PubMed
- For lead references, see:
(a) J. H. Birkinshaw, A. R. Chambers and H. Raistrick, Biochem. J., 1942, 36, 242–251 CrossRef CAS PubMed
-
(a) J. Davison, A. al Fahad, M. Cai, Z. Song, S. Y. Yehia, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7642–7647 CrossRef CAS PubMed
-
(a) F.-C. Yu, B. Zhou, H. Xu, Y.-M. Ki, J. Lin, S.-J. Yan and Y. Shen, Tetrahedron, 2015, 71, 1036–1044 CrossRef CAS
- For a prior example where purified oxidopyrylium dimer was advantageous, see: M. P. D'Erasamo, C. Meck, C. A. Lewis and R. P. Murelli, J. Org. Chem., 2016, 81, 3744–3751 CrossRef PubMed
- G. L. Beilhartz, M. Ngure, B. A. Johns, F. DeAnda, P. Gerondelis and M. Götte, J. Biol. Chem., 2014, 289, 16270–16277 CrossRef CAS PubMed
- S. Chung, D. M. Himmel, J. K. Jiang, K. Wojtak, J. D. Bauman, J. W. Rausch, J. A. Wilson, J. A. Beutler, C. J. Thomas, E. Arnold and S. F. J. Le Grice, J. Med. Chem., 2011, 54, 4462–4473 CrossRef CAS PubMed
- B. Zhang, M. P. D'Erasmo, R. P. Murelli and E. Gallicchio, ACS Omega, 2016, 1, 435–447 CrossRef CAS PubMed
-
(a) Y. Hu, X. Cheng, F. Cao, A. Huang and J. E. Tavis, Antiviral Res., 2013, 99, 221–229 CrossRef CAS PubMed
- J. E. Tavis, H. Wang, A. E. Tollefson, B. Ying, M. Korom, X. Cheng, F. Cao, K. L. Davis, W. S. M. Wold and L. A. Morrison, Antimicrob. Agents Chemother., 2014, 58, 7451–7461 CrossRef PubMed
- For some lead reviews, see:
(a) J. D. Baines, Trends Microbiol., 2011, 19, 606–613 CrossRef CAS PubMed
- S. S. Sigamani, H. Zhao, Y. N. Kamau, J. D. Baines and L. Tang, J. Virol., 2013, 87, 7140–7148 CrossRef CAS PubMed
- D. M. Himmel, K. A. Maegley, T. A. Pauly, J. D. Bauman, K. Das, C. Dharia, A. D. Clark Jr., K. Ryan, M. J. Hickey, R. A. Love, S. H. Hughes, S. Bergqvist and E. Arnold, Structure, 2009, 17, 1625–1635 CrossRef CAS PubMed
- J. L. Banks, H. S. Beard, Y. Cao, A. E. Cho, W. Damm, R. Farid, A. K. Felts, T. A. Halgren, D. T. Mainz, J. R. Maple, R. Murphy, D. M. Philipp, M. P. Repasky, L. Y. Zhang, B. J. Berne, R. A. Friesner, E. Gallicchio and R. M. Levy, J. Comput. Chem., 2005, 26, 1752–1780 CrossRef CAS PubMed
-
Schrödinger Release 2016-3, Glide, Schrödinger, LLC, New York, NY, 2016 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C7OB02453C |
| This journal is © The Royal Society of Chemistry 2018 |