DOI:
10.1039/C7NJ03305B
(Paper)
New J. Chem., 2018,
42, 483-488
Synergistic effects of Zn and Pd species in TiO2 towards efficient photo-reduction of CO2 into CH4†
Received
1st September 2017
, Accepted 26th November 2017
First published on 27th November 2017
Abstract
A Zn and Pd co-modified TiO2 photocatalyst exhibits remarkably enhanced photocatalytic activity upon reduction of CO2 into CH4. Owing to the synergistic effect of unique O–Zn–Cl and O–Pd–O species, TiO2–PdX–ZnY presents enhanced visible absorption and efficient charge carrier separation. It can be deduced from theoretical calculations and experimental results that the energy levels of O–Zn–Cl and O–Pd–O satisfy the redox potential of CO2 reduction, improving the photocatalytic performance. These results suggest that introducing multi-surface species to produce synergetic effects is a feasible route for efficient photocatalysis.
Introduction
Photo-reduction of CO2 into CH4via semiconductor photocatalysis has been investigated by many researchers since it uses solar light to convert CO2 into a valuable carbon fuel, methane.1–5 The mechanism of photo-reduction of CO2 into CH4 is explained as follows:6–13
| ˙OH + H2O + 3h+ → O2 + 3H+ |
| | | 2H2O + 4h+ → O2 + 4H+E0redox = 0.82 V vs. NHE | (2) |
| 2CO2 + 4e− → 2CO + O2E0redox = −0.12 V vs. NHE |
| ˙C + 4e− + 4H+ → CH4E0redox = 0.13 V vs. NHE |
| | | CO2 + 8e− + 8H+ → CH4 + 2H2O E0redox = −0.24 V vs. NHE | (3) |
Electrons and holes are excited under proper irradiation (eqn (1)). These charge carriers would further react with surface adsorbed CO2 and H2O molecules to generate CO and carbon radicals (˙C), which would further react with H+ to produce the final product, CH4 (eqn (2) and (3)). Furthermore, it is considered as necessary for highly active catalysts that the position of the conduction band and the valence band should satisfy the redox potential discussed above.
TiO2 is one of the most promising photocatalysts for the photoreduction of CO2 into CH4, due to its good performance and stability.2,6,8,14–18 However, the catalytic application of TiO2 is still limited because of its large band-gap, its rapid recombination of charge carriers, and especially the mismatch of its redox potential for the photoreduction of CO2 into CH4. Its modification with metal or nonmetal elements is considered as an effective method to enhance its visible response and separate the charge carriers as well as design and adjust the energy level of the photocatalyst, leading to full use of the photogenerated charge carriers. It is found that introducing Zn or Mn ions into the TiO2 system would create new doping energy levels below the conduction band, extending the response into the visible region and separating the charge carriers.19,20 Moreover, it is also noted that introducing Cu, C and Pd species into the TiO2 system would enhance the visible response and generate more charge carriers by forming new energy levels above the valence band.19,21
TiO2 doped or modified with Pd22–27 has been investigated to improve the photocatalytic performance recently. Electrons and holes would transfer to the surface to participate in the photo-reduction reaction more easily and rapidly via these doping energy levels. In our previous work, it was found that co-modified TiO2 with Pd and other elements could show a strong visible response and separate charge carriers efficiently,28–30 exhibiting remarkable catalytic performance. Moreover, the existence of unique Zn species on the TiO2 surface is also confirmed.23
To date, for TiO2 co-modified with Pd and other elements the band structure, its relationship with the redox potential for the reduction of CO2 into CH4, and the behaviour of the photogenerated charge carriers are still not investigated. Furthermore, the synergistic effect of Pd with other elements and their relationships with the visible response, separation of charge carriers and redox ability as well as the photocatalytic activity of the photocatalyst still need further investigation. And the photocatalytic mechanism should be discussed in detail.
In this work, Pd and Zn co-modified TiO2 exhibited remarkable photocatalytic activity. This paper focuses on the synergistic effect of Pd and Zn species on the surface and their influence on the band structure, visible response, separation of the photogenerated charge carriers and photo-catalytic activity in detail. The photocatalytic mechanism is also investigated for the reduction of CO2 into CH4.
Experimental
Synthesis of TiO2–PdX–ZnY
All chemicals used in this work were of analytical grade. At room temperature, different amounts of PdCl2 solution, Zn(NO3)2 and 1 mL of HCl solution (12 mol L−1) were dissolved in 40 mL of ethanol. Under violent stirring for 30 min, 12 mL of Ti(OC4H9)4 and 1 mL of deionized water were added dropwise (pH = 0.5). The mixed solution was stirred until TiO2 gel is formed. After 24 hours of aging, the TiO2 gel was heated at 373 K for 10 h and calcined at 723 K for 2.5 h. These synthesized samples are denoted as TiO2–PdX–ZnY (X = 1.5–3%; Y = 3–7%), where X and Y stand for the nominal molar ratio of Zn2+ and Pd2+ to Ti4+ (Zn/Ti and Pd/Ti), respectively. Pure TiO2, 5% Zn modified TiO2 (TiO2–Zn) and 1.5% Pd modified TiO2 (TiO2–Pd) were synthesized by the same process just without PdCl2 and/or Zn2(NO3)2.
Characterizations
The Raman spectrum was acquired on a Renishaw in Via Raman microscope with the 785 nm line of a Renishaw HPNIR 785 semiconductor laser. The BET surface areas were obtained by measuring nitrogen adsorption–desorption isotherms at 77 K (Micromeritics Automatic Surface Area Analyzer Gemini 2360, Shimadzu), after degassing at 453 K. XPS was performed with an ESCA Lab 220i-XL spectrometer by using an unmonochromated Al Ka X-ray source (148.6 eV). All spectra have been calibrated using the adventitious C1s peak at 284.8 eV. Diffuse reflectance UV-vis absorption spectra (UV-vis DRS) were obtained using a UV-vis spectrometer (U-4100, Hitachi). Photoluminescence (PL) spectra were measured on a spectrometer (Spex 1702), a photomultiplier tube (PMT, Hamamatsu R943), a lock-in amplifier, and a computer for data processing using the 325 nm line of a nano-second Nd:YAG laser (NL303G) as the excitation source. Surface photovoltage spectra (SPS) were acquired on a solid-junction photovoltaic cell of ITO/sample/ITO equipped with a lock-in amplifier (Model SR830, DSP) and synchronized with a light chopper. Monochromatic light was obtained by passing light from a 500 W xenon lamp (CHF-XQ 500W) through a double prism monochromator (WDG30-2).
Photo-reduction of CO2
The photoreduction of CO2 into CH4 is used to estimate the performance of the photocatalyst. A 500 W spherical Xenon arc lamp (Philips, Belgium, 35 mW cm−2, 290–800 nm) was applied as the light source in this experiment. There was one glass sheet (9.4 cm2) placed at the bottom of the sealed reaction vessel (410 mL). About 150 mg of the as-prepared samples was tiled on the glass. The light beam was vertical to the glass and the lamp was 10 cm away from the surface of the glass. The reaction vessel was filled with CO2 gas (99.99%) for 45 min before it was sealed. Then the reaction mixture was injected with 2 mL of water. About 0.4 mL of the gas in this vessel was extracted every 120 min. A gas chromatograph (Techcomp GC-7890F, equipped with a 1 m × 3 mm TDX-01 packed column and a flame ionization detector (FID)) was used to detect the concentration of the produced CH4 and CO. The relative humidity and temperature in the reactor during the photosynthesis process were maintained as 80% and 30 °C.
Results and discussion
The photo-reduction of CO2 into CH4 under Xe lamp irradiation is applied to estimate the catalytic performance of the obtained TiO2-based photocatalysts, as shown in Fig. 1, and Table 1. CO is the intermediate product (Fig. S1 and Table S1, ESI†) and CH4 is the final product. There is no other gas product detected, such as H2 and CH3OH. After 8 h of irradiation, only 0.350 μmol of CH4 is detected for pure TiO2. TiO2–Zn exhibited a finite photocatalytic activity and 0.851 μmol of CH4 was generated. The photocatalytic performance of TiO2–Pd was enhanced effectively and 3.59 μmol of CH4 was generated. After the introduction of Zn and Pd into TiO2, the TiO2–PdX–ZnY (X = 1.5–3%; Y = 3–7%) samples exhibited remarkably enhanced photocatalytic activity. Furthermore, the TiO2–Pd1.5%–Zn5% sample represented the highest catalytic performance of all photocatalysts and about 7.99 μmol of CH4 was detected, which is almost twice that for TiO2–Pd and twenty times higher than that for pure TiO2. The results indicated that the photocatalytic activity for the Zn and Pd co-modified TiO2 photocatalyst was enhanced remarkably due to the introduction of both Zn and Pd into TiO2. In addition, it was found from XPS that the amount of C before and after the photocatalytic reaction was 15.34% and 18.63%, respectively, suggesting that the surface absorbed carbon in the photocatalyst did not react with CO2 to generate CO.
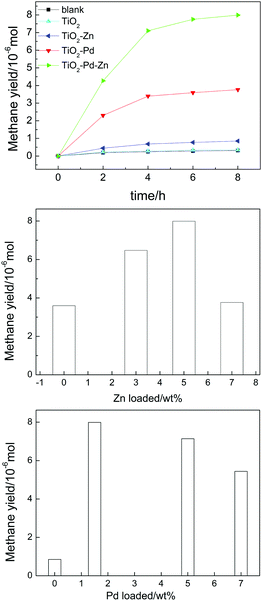 |
| | Fig. 1 Amount of CH4 generation for TiO2, TiO2–Pd, TiO2–Zn and TiO2–Pd1.5%–Zn5% (a), TiO2–Pd1.5%–ZnY% (b) and TiO2–PdX–Zn5% (c) under Xe lamp irradiation after 8 h. | |
Table 1 Photocatalytic activity of pure TiO2, TiO2–Zn, TiO2–Pd and TiO2–PdX–ZnY (X = 1.5–3%; Y = 3–7%) samples under Xe lamp irradiation after 8 h
| Sample |
CH4 (10−6 mol) |
Specific photocatalytic activity (10−6 mol g−1 h−1)b |
|
Blank is the photolysis of CO2.
Specific photocatalytic activity of CH4, CH4 generation amount per hour per unit mass of the catalyst. The experimental error in the photocatalytic results is <5% based on triplicate runs.
|
| Blanka |
0.31 ± 0.01 |
— |
| TiO2(P25) |
0.36 ± 0.02 |
0.30 ± 0.02 |
| TiO2 |
0.35 ± 0.03 |
0.29 ± 0.02 |
| TiO2–Zn |
0.85 ± 0.02 |
0.71 ± 0.02 |
| TiO2–Pd |
3.77 ± 0.47 |
3.14 ± 0.39 |
| TiO2–Pd1.5%–Zn3% |
6.48 ± 0.27 |
5.40 ± 0.23 |
| TiO2–Pd1.5%–Zn5% |
7.99 ± 0.28 |
6.66 ± 0.23 |
| TiO2–Pd1.5%–Zn7% |
3.60 ± 0.35 |
3.00 ± 0.29 |
| TiO2–Pd5%–Zn5% |
7.14 ± 0.45 |
5.95 ± 0.38 |
| TiO2–Pd7%–Zn5% |
5.44 ± 0.73 |
4.53 ± 0.61 |
The structure and the existing states of Pd and Zn were investigated by the Raman, XRD and XPS spectra for TiO2, TiO2–Zn, TiO2–Pd and TiO2–PdX–ZnY samples. For the Raman spectra (Fig. 2 and Fig. S2, ESI†), all samples showed typical characteristic bands and the vibrational modes of anatase.31 In addition, for TiO2–Zn as well as TiO2–PdX–ZnY samples (Fig. S2, ESI†), two additional weak Raman peaks around 256 cm−1 and 333 cm−1 were observed, ascribed to the stretching modes of Zn–Cl and Zn–O bonds, respectively.20 XRD results for all samples (Fig. S3 and Table S2, ESI†) also confirm that only anatase exists for these samples, which is in good agreement with the Raman spectrum mentioned above. Furthermore, after the introduction of Pd and Zn, the specific surface areas (BET) increase in the order of TiO2 < TiO2–Zn < TiO2–Pd < TiO2–Pd–Zn. The crystallite sizes decrease in the order of TiO2–Pd–Zn < TiO2–Zn < TiO2–Pd < TiO2. The detailed existing states of Zn and Pd are investigated by XPS.
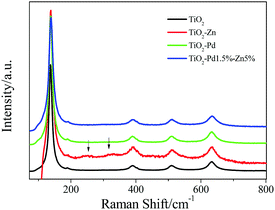 |
| | Fig. 2 Raman spectra of TiO2, TiO2–Zn, TiO2–Pd and TiO2–Pd1.5%–Zn5%. | |
Fig. 3 shows the XPS spectra of TiO2, TiO2–Pd, TiO2–Zn and TiO2–Pd1.5%–Zn5%. For the Pd 3d spectra (Fig. 3A), the peaks of Pd 3d5/2 for TiO2–Pd and TiO2–Pd1.5%–Zn5% locate at about 336.7 eV, attributed to the O–Pd–O structure on the TiO2 surface (one Pd2+ ion linked with two unsaturated O2− ions).19,32 For XPS Cl 2p spectra (Fig. 3B), the peak centered at about 198.1 eV for TiO2 and TiO2–Pd is ascribed to Cl 2p of the O–Ti–Cl species on the TiO2 surface. For the TiO2–Zn and TiO2–Pd1.5%–Zn5% samples, the peaks centered at about 198.9 eV and 198.7 eV respectively, are between those of TiCl4 (198.2 eV) and ZnCl2 (199.7 eV).9 Furthermore, as shown in Fig. 3C, the center of the Zn 2p3/2 peak for the Zn 2p spectra of TiO2–Zn (1022.1 eV) and TiO2–Pd–Zn (1022.0 eV) locates between those of ZnO (1021.5 eV)33 and ZnCl2 (1022.5 eV).34 This indicates that one Zn2+ ion links with one unsaturated O atom and one Cl− ion to exist as O–Zn–Cl species.20,34 Moreover, compared with TiO2 (2.01%) and TiO2–Pd (2.36%), the atom percentage calculated from the XPS also increases remarkably for TiO2–Zn (6.49%) and TiO2–Pd–Zn (5.25%). Hence, it is concluded that unique O–Pd–O and O–Zn–Cl species are formed on the surface of TiO2.
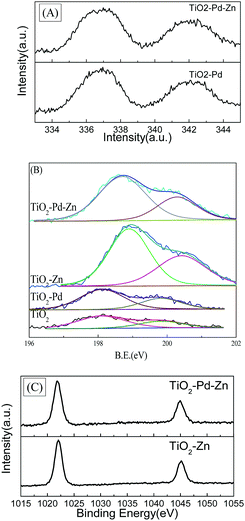 |
| | Fig. 3 (A) XPS Pd 3d spectra of TiO2–Pd and TiO2–Pd1.5%–Zn5%; (B) Cl 2p spectra of TiO2, TiO2–Pd, TiO2–Zn and TiO2–Pd–Zn; (C) Zn 2p of TiO2–Pd and TiO2–Pd1.5%–Zn5%. | |
To determine the band structure of TiO2–PdX–ZnY, the valence bands of XPS spectra and absorption spectra are shown in Fig. 4. For the XPS valence band spectra (Fig. 4A), the energy levels in all samples are aligned by the Fermi level of the XPS instrument (4.1 eV).35 The BE of the onset edge for O 2p revealed the gap between the valence band top and the Fermi level (Ef). The onset edge of the valence band top for TiO2 is at about +2.7 eV (+2.3 V, vs. NHE). The onset edge for TiO2–Zn almost remains unchanged compared with TiO2, implying that the energy level of O–Zn–Cl species is far from the valence band of TiO2. It is found from the theoretical calculation (Fig. S4, ESI†) that a new energy level corresponding to O–Zn–Cl species occurred below the conduction band of TiO2, which mainly consists of Zn 4s and Cl 3p orbitals. Moreover, for TiO2–Pd and TiO2–Pd–Zn, a small hump of about +2.1 eV (+1.7 eV, vs. NHE) is caused by the introduced Pd 4d orbital corresponding to the O–Pd–O species, whose energy level can be estimated to be at 0.6 eV above the valence band of TiO2. The theoretical simulation (Fig. S4, ESI†) also confirmed that new energy levels, composed of the Pd 4d orbital and hybridized with O 2p and Ti 3d states, occurred above the valence band of TiO2 for TiO2–Pd.
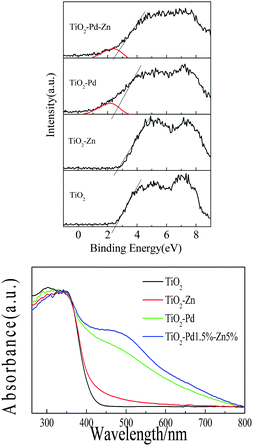 |
| | Fig. 4 (A) XPS valence band spectra, and (B) absorption spectra of TiO2, TiO2–Zn, TiO2–Pd and TiO2–Pd1.5%–Zn5%. | |
The absorption spectra of all samples are plotted in Fig. 4B. TiO2 shows strong absorption in the UV region caused by the band-to-band transition. The band gap for TiO2 is estimated to be 3.1 eV, as the onset edge is at about 400 nm. Weak absorption centered at about 450 nm from 400 nm to 600 nm for TiO2–Zn is observable. Electrons in the valence band can be excited to the energy level of O–Zn–Cl species next to the conduction band. The stronger visible response is observed for TiO2–Pd, as the electrons from O–Pd–O species can be excited to the conduction band of TiO2. Moreover, as we expect, owing to the synergistic effect of O–Pd–O and O–Zn–Cl species, TiO2–PdX–ZnY represents the strongest absorption in the visible region (Fig. 4B and Fig. S3, ESI†), and the absorption can be further improved by increasing the amount of Pd and Zn. These results indicate that introducing Zn into the TiO2–Pd system could lead to the synergetic effect of Pd and Zn, which may enhance the visible response, and match the band structure with the redox potential of photo-reduction of CO2 into CH4, resulting in enhanced catalytic activity.
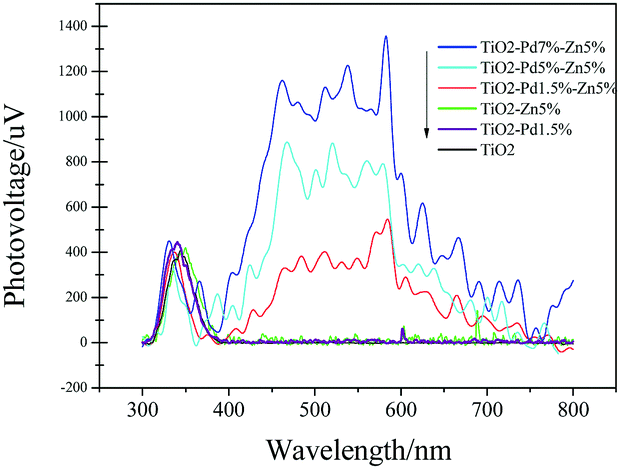
To investigate the behaviour of charge carriers, photoluminescence spectra (PL), time resolved PL and surface photovoltaic spectra (SPS) were acquired, as shown in Fig. 5 and 6 and Table 2. For the photocatalyst, the electrons fell into the oxygen vacancies from the conduction band through a nonirradiative process (τ1) and recombined with holes via an irradiative process (τ2), resulting in fluorescence emission. Hence, quenching of the PL intensity and prolonged life-time of charge carriers indicate efficient suppression of the recombination for charge carriers. Fig. 5 shows the PL spectra of TiO2, TiO2–Zn, TiO2–Pd and TiO2–PdX–ZnY. It is observed from Fig. 5A that the emission intensity of TiO2–Zn is weakened compared with TiO2, owing to the surface energy level of O–Zn–Cl species. For TiO2–Pd (Fig. 5B), the PL intensity is quenched significantly because the holes transfer from the valence band to the O–Pd–O species. And the PL emission for TiO2–PdX–ZnY is further quenched with the increase of Zn or Pd owing to the contribution from both the Zn and Pd species (Fig. 5A and B). Moreover, the τ1 and τ2 values related to the nonirradiative and irradiative processes are shown in Table 2. The τ2 values for TiO2–Zn and TiO2–Pd are greater than that for TiO2, as the electrons could transfer from the conduction band of TiO2 to the levels of O–Zn–Cl species or the holes could move from the valence band to the energy levels of O–Pd–O species. The τ2 values for TiO2–Pd1.5%–Zn5% samples are greater than those of TiO2–Zn and TiO2–Pd due to the synergistic effect of Pd and Zn species. These results suggest that the photo-generated electrons and holes were separated more efficiently for Zn and Pd co-modified TiO2 samples than TiO2–Zn and TiO2–Pd.
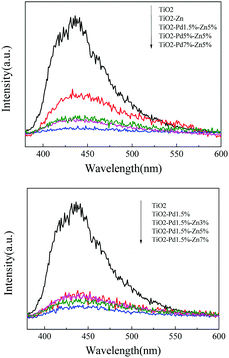 |
| | Fig. 5 Photoluminescence spectra (PL) of TiO2, TiO2–Zn, TiO2–Pd, and TiO2–PdX–Zn5% (A) and TiO2–Pd1.5%–ZnY (B). | |
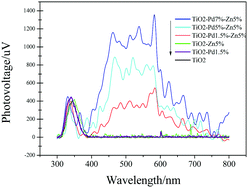 |
| | Fig. 6 Surface photovoltaic spectra (SPS) of TiO2, TiO2–Zn, TiO2–Pd and TiO2–PdX–ZnY. | |
Table 2 Time constant τ1 and τ2 through double exponential decay fitting for TiO2, TiO2–Zn, TiO2–Pd, and TiO2–Pd1.5%–Zn5%
|
|
TiO2 |
TiO2–Zn |
TiO2–Pd |
TiO2–Pd1.5%–Zn5% |
|
τ
1 (ns) |
0.295 |
0.298 |
0.294 |
0.291 |
|
τ
2 (ns) |
3.059 |
3.876 |
4.038 |
4.382 |
As shown in Fig. 6, SPS reveals the separation behaviours of photogenerated charge carriers. The higher light response (peak intensity) usually indicates that more charge carriers are separated. For all samples, the response peaks around 350 nm are caused by band–band transition. TiO2 shows no SPS response in the visible region (400 nm to 800 nm). And TiO2–Zn and TiO2–Pd exhibit poor SPS response in the visible region compared with TiO2. This indicates that the photo-generated charge carriers were not separated efficiently from the surface of the catalyst under visible irradiation, when only Zn or Pd species were introduced into the TiO2 system. Moreover, the TiO2–PdX–ZnY samples exhibited a significant light response in the visible region compared to the others. With increasing Pd content, the light response of TiO2–PdX–Zn5% is further enhanced, which may be attributed to the synergistic effect of O–Zn–Cl and O–Pd–O species. This suggests that more electrons and holes were generated and separated for the TiO2–PdX–ZnY samples than the TiO2–Pd or TiO2–Zn samples, respectively.
According to the SPS results (Fig. 6), the synergistic effect of Pd and Zn species on the surface can be analyzed by the schematic band structure of TiO2–PdX–ZnY samples (Fig. 7). TiO2 shows no visible response and the recombination rates of holes and electrons are quite high. For TiO2–Zn, the weaker SPS response in the visible region is assigned to the limited visible absorption in the visible region. Stronger visible absorption is observed for TiO2–Pd, caused by the O–Pd–O species. Moreover, the SPS response in the visible region is poor, due to the rapid recombination of charge carriers, as shown in Fig. 7. For TiO2–PdX–ZnY, the higher SPS light response in the visible region indicates the more efficient generation and separation of photogenerated charge carriers, owing to the synergistic effect of Pd and Zn species on the TiO2 surface. Electrons were generated from the valence band to the energy level of Zn species. Furthermore, the electrons in the conduction band arising from Pd species can also transfer to the levels of O–Zn–Cl species, instead of recombining with holes in the valence band, separating photogenerated charge carriers efficiently. Therefore, for TiO2–PdX–ZnY, more charge carriers were excited, separated and transferred to the surface of the photocatalyst than the TiO2 samples modified with only Pd or Zn on the surface. When the electrons and holes migrate to the surface, the Zn and Pd surface species may act as reactive sites.
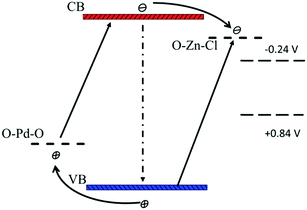 |
| | Fig. 7 Schematic band structure of TiO2–PdX–ZnY as well as the photocatalytic mechanism (not drawn to scale). | |
Because the energy level of O–Zn–Cl (−0.45 V, vs. NHE) locates above the reduction–oxidation potential of reaction (3) (−0.24 V, vs. NHE) and the level of O–Pd–O (1.7 V, vs. NHE) is below the potential of reaction (2) (+0.8V, vs. NHE) respectively, these photo-generated carriers on the surface would participate in the catalytic reaction upon reduction of CO2 into CH4. According to the discussion above, owing to the synergistic effect of O–Zn–Cl and O–Pd–O species on the TiO2 surface, the visible response was improved significantly and charge carriers were separated effectively. Moreover, the band structure for TiO2–PdX–ZnY photocatalysts match preferably with the redox potential upon the photoreduction of CO2 into CH4, because of the energy levels of the O–Pd–O and O–Zn–Cl species. As a result, more photogenerated holes and electrons could be involved in the photocatalytic reduction, leading to a much higher photocatalytic activity for TiO2–PdX–ZnY than for TiO2–Pd and TiO2–Zn.
Conclusions
A series of highly reactive photocatalysts, TiO2–PdX–ZnY, were synthesized and exhibited significantly improved photo-reduction activity of CO2 into CH4. Because of the synergetic effect of surface O–Zn–Cl and O–Pd–O species, visible absorption was enhanced significantly and charge carriers were separated. Because the energy level of surface species is close to the redox potential of photo-reduction, more electrons and holes are enriched in the reactive sites (O–Zn–Cl and O–Pd–O surface species) and further participate in photo-reduction, leading to a remarkably improved photocatalytic activity for TiO2–PdX–ZnY upon photoreduction of CO2 into CH4.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 51372120) and China Postdoctoral Science Foundation (no. 2017M611149). We thank Prof. Yihong Ding from State Key Lab of Theoretical and Computational Chemistry, Jilin University for helping us to carry out the theoretical calculation.
Notes and references
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- K. F. Li, X. Q. An, K. H. Park, M. Khraisheh and J. W. Tang, Catal. Today, 2014, 224, 3–12 CrossRef CAS.
- L. Yuan and Y. J. Xu, Appl. Surf. Sci., 2015, 342, 154–167 CrossRef CAS.
- O. Ola and M. M. Maroto-Valer, J. Photochem. Photobiol., C, 2015, 24, 16–42 CrossRef CAS.
- S. Bai, W. Yin, L. Wang, Z. Li and Y. Xiong, RSC Adv., 2016, 6, S7446–S7463 Search PubMed.
- N. M. Dimitrijevic, B. K. Vijayan, O. G. Poluektov, T. Rajh, K. A. Gray, H. He and P. Zapol, J. Am. Chem. Soc., 2011, 133, 3964–3971 CrossRef CAS PubMed.
- Q. Liu, Y. Zhou, Z. Tian, X. Chen, J. Gao and Z. Zou, J. Mater. Chem., 2012, 22, 2033–2038 RSC.
- S. Xie, Y. Wang, Q. Zhang, W. Fan, W. Deng and Y. Wang, Chem. Commun., 2013, 49, 2451–2453 RSC.
- Y. Yan, Y. Yu, D. Wu, Y. Yang and Y. Cao, Nanoscale, 2015, 8, 949–958 RSC.
- Y. Yu, L. Guo, H. Cao, Y. Lv, E. Wang and Y. Cao, Sep. Purif. Technol., 2015, 142, 14–17 CrossRef CAS.
- E. Karamian and S. Sharifnia, J. CO2 Util., 2016, 16, 194–203 CrossRef CAS.
- K. Koci, L. Obalova and O. Solcova, Chemical and Process Engineering-Inzynieria Chemiczna I Procesowa, 2010, 31, 395–407 CAS.
- S. T. Seng, L. Zou and E. Hu, Catal. Today, 2008, 131, 125–129 CrossRef.
- K. Bhattacharyya, A. Danon, B. K. Vijayan, K. A. Gray, P. C. Stair and E. Weitz, J. Phys. Chem. C, 2013, 117, 12661–12678 CAS.
- V. V. Galvita, H. Poelman, C. Detavernier and G. B. Marin, Appl. Catal., B, 2015, 164, 184–191 CrossRef CAS.
- C. Wang, R. L. Thompson, J. Baltrus and C. Matranga, J. Phys. Lett., 2009, 1, 48–53 Search PubMed.
- J. Low, B. Cheng and J. Yu, Appl. Surf. Sci., 2017, 392, 658–686 CrossRef CAS.
- L. Liu and Y. Li, Aerosol Air Qual. Res., 2014, 14, 453–469 CAS.
- Y. Yan, Y. Yu, S. Huang, Y. Yang, X. Yang, S. Yin and Y. Cao, J. Phys. Chem. C, 2017, 1089–1098 CAS.
- S. Yan, Y. Yu, Y. Gu, Y. Liu and Y. Cao, Sep. Purif. Technol., 2016, 171, 118–122 CrossRef CAS.
- Y. Yan, Y. Yu, C. Cao, S. Huang, Y. Yang, X. Yang and Y. Cao, CrystEngComm, 2016, 18, 2956–2964 RSC.
- M. Bowker, D. James, P. Stone, R. Bennett, N. Perkins, L. Millard, J. Greaves and A. Dickinson, J. Catal., 2003, 217, 427–433 CrossRef CAS.
- Y. Yu, J. Wang, W. Li, W. Zheng and Y. Cao, CrystEngComm, 2015, 17, 5074–5080 RSC.
- F. N. Sayed, O. Jayakumar, R. Sasikala, R. Kadam, S. R. Bharadwaj, L. Kienle, U. Schürmann, S. R. Kaps, R. Adelung and J. Mittal, J. Phys. Chem. C, 2012, 116, 12462–12467 CAS.
- A. T. Kuvarega, R. W. M. Krause and B. B. Mamba, J. Phys. Chem. C, 2011, 115, 22110–22120 CAS.
- Q. Li, Y. W. Li, P. Wu, R. Xie and J. K. Shang, Adv. Mater., 2008, 20, 3717–3723 CrossRef CAS.
- Q. Li, R. Xie, E. A. Mintz and J. K. Shang, J. Am. Ceram. Soc., 2007, 90, 3863–3868 CAS.
- Y. Yu, E. Wang, J. Yuan and Y. Cao, Appl. Surf. Sci., 2013, 273, 638–644 CrossRef CAS.
- Y. Cao, T. He, L. Zhao, E. Wang, W. Yang and Y. Cao, J. Phys. Chem. C, 2009, 113, 18121–18124 CAS.
- Y. Cao, Y. Yu, P. Zhang, L. Zhang, T. He and Y. Cao, Sep. Purif. Technol., 2013, 104, 256–262 CrossRef CAS.
- J. Wang, Y. Yu, S. Li, L. Guo, E. Wang and Y. Cao, J. Phys. Chem. C, 2013, 117, 27120–27126 CAS.
- Y. Yu, T. He, L. Guo, Y. Yang, L. Guo, Y. Tang and Y. Cao, Sci. Rep., 2015, 5(9561), 1–6 Search PubMed.
- W. Li, D. Wu, Y. Yu, P. Zhang, J. Yuan, Y. Cao, Y. Cao and J. Xu, Phys. E, 2014, 58, 118–123 CrossRef CAS.
- Y. Yu, Y. Gu, W. Zheng, Y. Ding and Y. Cao, J. Phys. Chem. C, 2015, 119, 28190–28193 CAS.
- Y. Cao, T. He, Y. Chen and Y. Cao, J. Phys. Chem. C, 2010, 114, 3627–3633 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nj03305b |
| ‡ Both authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |

 *a
*a