
Rapid spheroid clearing on a microfluidic chip†
Tomas
Silva Santisteban
ab,
Omid
Rabajania
ab,
Iana
Kalinina
ab,
Stephen
Robinson
ab and
Matthias
Meier
 *ab
*ab
aMicrofluidic and Biological Engineering, IMTEK - Department of Microsystems Engineering, University of Freiburg, Georges-Koehler-Allee 103, 79110 Freiburg, Germany. E-mail: matthias.meier@imtek.de
bBIOSS - Centre for Biological Signalling Studies, University of Freiburg, Germany
First published on 24th November 2017
Abstract
Spheroids are three-dimensional (3D) cell cultures that aim to bridge the gap between the use of whole animals and cellular monolayers. Microfluidics is regarded as an enabling technology to actively control the chemical environment of 3D cell cultures. Although a wide variety of platforms have been developed to handle spheroid cultures, the development of analytical systems for spheroids remains a major challenge. In this study, we engineered a microfluidic large-scale integration (mLSI) chip platform for tissue-clearing and imaging. To enable handling and culturing of spheroids on mLSI chips, with diameters within hundreds of microns, we first developed a general rapid prototyping procedure, which allows scaling up of the size of pneumatic membrane valves (PMV). The presented prototyping method makes use of milled poly(methylmethacrylate) (PMMA) molds for obtaining semi-circular microchannels with heights up to 750 μm. Semi-circular channel profiles are required for the functioning of the commonly used PMVs in normally open configuration. Height limits to tens of microns for this channel profile on photolithographic molds have hampered the application of 3D tissue models on mLSI chips. The prototyping technique was applied to produce an mLSI chip for miniaturization, automation, and integration of the steps involved in the tissue clearing method CLARITY, including spheroid fixation, acrylamide hydrogel infiltration, temperature-initiated hydrogel polymerization, lipid extraction, and immuno-fluorescence staining of the mitochondrial protein COX-IV, and metabolic enzyme GAPDH. Precise fluidic control over the liquids in the spheroid culturing chambers allowed implementation of a local hydrogel polymerization reaction, exclusively within the spheroid tissue. Hydrogel-embedded spheroids undergo swelling and shrinkage depending on the pH of the surrounding buffer solution. A pH-jump from 8.5 to 5.5 shrinks the hydrogel-embedded spheroid volume by 108% with a rate constant of 0.36 min−1. The process is reversible upon increasing the pH, with the rate constant for the shrinkage being −0.12 min−1. Repetitive cycling of the pH induces an osmotic flow within the hydrogel-embedded spheroid. Thirty cycles, performed in a total time interval of 10 minutes on-chip, reduced the clearing time of a hydrogel-embedded spheroid (with a diameter of 200 μm) from 14 days to 5 hours. Therefore, we developed a physicochemical method to decrease the clearing time of hydrogel-embedded tissues. While the osmotic pump mechanism is an alternative to electrophoretic forces for decreasing tissue clearing times, the integration of the CLARITY method on chip could enable high throughput imaging with 3D tissue cultures.
Introduction
Spheroids are attractive three-dimensional (3D) cell culture model systems for integration with microfluidic chip applications.1,2 The umbrella term “spheroid” describes microscale spherical cell clusters formed by self-assembly, which are used to study in vitro organ and cancer development,3 tissue regrowth,4 and/or function of the tissue in in vivo counterparts.5 In contrast to 2D cell cultures, cells within spheroids form increased cell–cell junctions and extracellular matrix. Furthermore, peripheral cells of a spheroid are in direct contact with the cell medium and can form epithelial cell structures, whereas internal cells of the spheroid are encapsulated and experience varied nutrient gradients.6 Therefore, spheroids can more accurately recapitulate the in vivo microenvironment than 2D cell cultures, and also offer the possibility to study signaling across cell barriers. Concomitantly, the simplicity of this cell culture technique allows for multiple experimental replicates to be performed in a reduced amount of time compared to animal studies.7 This has led to the widespread implementation of spheroid-based technologies in pharmaceutical research.Spheroid cultures are variable in size and shape, depending on their cell type composition, heterogeneity, and developmental stage. Using standardized hanging-drop,8 non-adhesive surfaces,9 or suspension culture10 technologies, spheroids can be formed with defined diameters from tens to hundreds of microns. Owing to their size, spheroids are ideally suitable for integration on microfluidic chip platforms. There are vast varieties of chip designs possible for the automation of spheroid formation,11,12 handling,12,13 culturing,14 and manipulation.15,16 Despite these achievements in microfluidic engineering, a major challenge continues to be the development of analytical systems for spheroids. In particular, we are confronted with a lack of simple imaging tools for analysis of biomolecules in deeper spheroid tissue layers that are compatible with microfluidic chip technologies. In general, fluorescence staining approaches are hampered by cell barriers. Densely packed lipid bilayers act as diffusion barriers for molecular probes. Therefore, biomolecular staining with fluorescently labeled antibodies or oligonucleotides leads to inhomogeneous staining in the deeper cell layers of 3D tissue models, and requires days for clearing of non-specifically bound probes. In addition, photons scatter at the lipid–aqueous interface of cell membranes, which limits the optical penetration of light into 3D spheroid cultures. Consequently, the utility of conventional microscopy is restricted to superficial areas, and only two-photon17 or light-sheet microscopy can reach a deeper layer of penetration.18 Both imaging approaches have been applied to analyze biomolecules in spheroids cultured on microfluidic chips; however, these technologies require sophisticated imaging equipment, can only characterize a few targets at a time, and are not applicable for high-throughput screens. Consequently, clinical studies with spheroids still rely on sectioning and subsequent immunohistochemistry analysis.
Recently, several tissue-clearing methods have been developed to facilitate imaging in deeper tissue layers without the need of sectioning.19 The most common method is known as clear, lipid-exchange, anatomically rigid imaging/immunostaining-compatible tissue hydrogel (CLARITY).20 In this protocol, cellular biomolecules, including proteins, nucleic acids, and small molecules, are cross-linked with each other and to a non-porous hydrogel. Lipids lacking functional groups for conjugation remain unbound and can be extracted using detergents from the hybrid hydrogel–tissue structure. The hydrogel mesh physically supports the tissue structure and further enables analysis of the tissue with immunofluorescence microscopy. Embedding of cells or tissues in hydrogels has led to novel biomolecular analytical methods such as expansion microscopy,21 in which swelling properties are exploited to increase the optical resolution for cellular structures. Unfortunately, CLARITY requires approximately 14 days, even for small tissue samples such as spheroids. The long clearing time accompanied by a multistep preparation protocol makes this method incompatible for higher throughput studies.
Mirofluidic large-scale integration technology (mLSI) has proven to automate and parallelize complex bioanalytical workflows for 2D cell cultures. However, the relatively large size of spheroids remains an obstacle for mLSI chip technology. The central fluid control elements incorporated in the mLSI chip technology are pneumatic membrane valves (PMVs). PMVs can be designed in either a normally open or closed configuration. The normally open configuration is predominately used due to its simpler production workflow and saver control operation. Unfortunately, for closing a fluidic microchannel with a PMVs in normally open configuration, requires that the flow channel exhibits a semi-circular channel profile. Production processes of rapid prototyping molds for the mLSI chips have only been established for microchannels up to 50 μm in height due to photoresist limitations.22 This problem has thus limited the application of mLSI technology to 2D cell cultures. Consequently, new production methods are required to transfer the multiplexing advantages of mLSI chip technology23,24 from 2D to 3D cell cultures or biopsies.
In order to integrate spheroid cell culturing and deep-tissue biomolecular imaging analytics on-chip, we developed a new rapid prototyping production process for mLSI chips. To overcome the size restriction for PMVs, we replaced the photolithography molds for polydimethylsiloxane casting with high-precision milled plastic molds. With the milled plastic molds, it is possible to fabricate PMVs closing fluidic channels with a ten-times larger height than possible with current PMVs. We next used the developed rapid prototyping process to engineer mClear-Chip, which exploits the scaled PMVs for integrating the CLARITY methodology for spheroid cultures. In contrast to CLARITY in batch experiments, the advances of fluidic control mechanisms are exploited to polymerize hydrogels only within the spheroid tissue structures rather than the surrounding solutions, which simplifies the washing of the samples. Furthermore, upon cycling the pH value in the surrounding hydrogel-embedded spheroids, it is possible to induce an osmotic pump process to reduce the amount of lipid extraction from the spheroids and thus substantially lower the tissue-clearing time from 10–14 days to 5 h.
Materials and methods
Spheroid formation
Adipose-derived stem cells (hASCs) were obtained from ZenBio (Research Triangle Park, NC, USA). Spheroids were generated using the agarose overlay technique.25 Each well of a 96-well plate was filled with 120 μl 4% agarose. Approximately 1500 cells in 100 μl growth medium were pipetted on top of the agar, which resulted in spheroids with a diameter between 200 ± 50 μm. Spheroids were formed during an overnight incubation at 37 °C on an orbital shaker at 600 rpm. hASC spheroids were then transferred with a pipette (20 μL) to the mLSI chip through the spheroid loading port, and the “macro valves” were engaged to enclose the spheroids within the device. The diameter of the spheroids was determined from brightfield images.Mold and connector fabrication
Silicon molds for PDMS soft-lithography were produced using a previously published procedure.26 In short, emulsion masks with a resolution of 50k dpi were commercially obtained (BVM maskshop, Oberhausen, Germany). All silicon molds used in this work consisted of two layers of photoresist. All flow microchannels on silicon substrates were patterned with AZ 40XT (MicroChemicals, Ulm, Germany) and were 120 × 25 μm (width, height). A semi-circular channel geometry for the fluidic channels was achieved by refloating the photoresist at 200 °C for 12 h. Control channels on the silicon were manufactured with SU-8 3025 (MicroChem, Newton, MA, USA) following the vendor's instructions. In general, control microchannels were 120 μm × 40 μm (width, height), except it is stated differently (e.g. the via element on the mold was 400 × 100 μm (width, height)). Silicon molds were coated with C4F8. The macro-valve on the mClear-Chip had a closing area of 500 × 1600 μm2 (width, height).The top PMMA layer, PMMA pressure adaptor, and positive PMMA mold for the top PDMS layer were milled on a Kern EVO Microtechnik (Kern, Eschenlohe, Germany). Square- and round-shaped microchannels were milled with a ball nose end mill (product no. VC2PSBPR0005) from Mitsubishi Material (Japan), and NSME230 from Gde Werkzeuge (Halver, Germany), respectively. The polarity of the PMMA mold for the upper PDMS layer was inverted by replicating a negative PDMS mold. For this, the PMMA mold was coated with (hydroxypropyl)methylcellulose (HPMC) to facilitate the release of PDMS.27 Liquid PDMS with a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of Sylgard 184 (Dow Corning, Midland, MI, USA) base material to the cross-linker was poured on top of the mold and then cured at 80 °C for 60 min. The release of a PMMA layer from a silicon mold is not possible, owing to the rigidity of both materials; therefore, the negative silicon mold was transferred into a PDMS mold by double casting.28
:1 ratio of Sylgard 184 (Dow Corning, Midland, MI, USA) base material to the cross-linker was poured on top of the mold and then cured at 80 °C for 60 min. The release of a PMMA layer from a silicon mold is not possible, owing to the rigidity of both materials; therefore, the negative silicon mold was transferred into a PDMS mold by double casting.28
Chip fabrication and assembly
To manufacture the upper PDMS layer of the mClear-Chip, the negative PDMS mold was treated with (hydroxypropyl)methyl cellulose (HPMC). Next, PDMS (5:1 ratio) was casted onto the HPMC-treated PDMS wafer mold to a thickness of about 1 mm and cured at 80 °C for 25 min. The lower PDMS layer was produced by spin-coating PDMS (ratio 20:1) to a thickness of 65 μm onto the silicon mold. At this height, the via element stood freely out of the PDMS film due to the hydrophilic C4F8 coating.29 The lower PDMS layer was baked for 15 min at 80 °C.
To assemble the layers of the mClear-Chip, the milled PMMA top layer was plasma-activated and then dip-coated with thermally activated 5% (v/v) (3-aminopropyl)triethoxysilane (3-APTES) solution for 1 min. The PMMA layer was blow-dried with nitrogen and kept at 65 °C on a hotplate for 10 min. Then, the PMMA plate and upper PDMS layer were plasma bonded, where the bond was strengthened at 80 °C for 5 min.30 After release of the negative PDMS mold, the upper PDMS layer was aligned to the lower PDMS layer. Bonding between the two PDMS layers was facilitated by backing the chip for 1 h at 80 °C after alignment (off-ratio PDMS binding). The layers were punctured with a 21-gauge needle to form interconnections. Finally, the assembled PDMS chip was plasma bonded to a glass carrier with a thickness of 0.17 mm (Brain Laboratories, USA). For a graphical summery of the chip production process see Fig. S1.† The microfluidic chip was controlled by a custom-made pressure system (for instructions, see Stanford Microfluidic Foundry webpage or the ref. 31).
The CLARITY flush program
The PDMS channel network was pre-treated with 1% BSA to block the channels. hASC spheroids were cultured in DMEM for 1 day on the chip before fixation. The culture medium was refreshed every hour. The spheroid fixation solution consisted of 1× PBS containing paraformaldehyde, polyacrylamide, and bisacrylamide (4/4/0.25 vol%) in PBS. The radical initiator VA-044 AZO was dissolved in phosphate buffered saline (1× PBS) at a concentration of 25 mg ml−1, which was 10 times higher than that in the original CLARITY protocol. To clear the spheroids, we used a solution of 0.14 M SDS in 0.2 M boric acid at pH 8.5. For the pH jump experiments, the clearing solution was exchanged with 0.05 M MES buffer at pH 5.5. All solutions were flushed with a flow pressure of 1 psi, which corresponded to a flow rate of 2 μl min−1 in the spheroid-trapping chamber of the chip. To ensure complete medium exchange, the flush time per chamber was 20 s, except for the introduction of the radical starter, which was 2 s.Spheroid imaging
Bright-field images were taken on a Zeiss Axio Observer with a 20× (Plan-Apochromat) objective. Images were acquired with LSM-I-NLO ZEISS LSM 880 Observer/Fast Airyscan combined with inverted microscope controlled by ZEN 2.3 software from Zeiss. We used LD-C-Apochromat 25×/0.8 Imm Korr DIC M27 (420852-9870) objective with water immersion. Cell nuclei in cleared spheroids were stained with DAPI (Sigma-Aldrich Co., St. Louis, MO, USA). The mitochondrial protein cytochrome c oxidase IV (COX IV) was detected with rabbit primary anti-COX IV antibody labeled with Alexa 647 dye (NEB, 7561). Spheroids were incubated with the corresponding antibodies for 8 h and then washed for 2 h with PBS (pH 7.2). The antibodies were post-fixated for 2 h with a 4% PFA solution. At last, X-Clarity (Logos Biosystems, USA) was introduced as mounting media to match the refractive index. Image analysis was performed with Matlab (Mathworks, USA) and ImageJ.Results
Scaling up pneumatic membrane valves on mLSI chips
A common obstacle encountered in the analysis of spheroid tissue on closed mLSI chip platforms is the mismatch of dimensions between the biological sample and the functional elements of the chip. Sizes of spheroids are in the order of several hundred micrometers, whereas the microchannels and commonly used PMVs in normally open configuration of mLSI chips are designed for sizes of tens of micrometers. PMVs in the normally open configuration can be produced in either a “push-up” or “push-down” configuration.32 The closing pressures depend on valve dimensions and the thickness of the membrane separating the channels for both configurations. Higher pressures are required to close “push-down” valves. In both configurations, the performance of these valves is dependent on the cross-sectional area between flow and control channel.33,34 For complete closing of a pneumatic membrane valve, the flow channels must show a semi-circular channel profile, which is one of the most limiting factors for up-scaling. Rounded channel profiles for microchannels on molds used for PDMS soft-lithography are obtained by refloating positive photoresists. AZ40XT or SPR 220 are commonly used to pattern the flow channel networks. However, at microchannel heights over 65 μm, the refloating process disrupts channel geometries, and creates channel offsets due to uncontrollable gravity flow of the resists. Therefore, the pneumatic membrane valves for mLSI chips are limited by the mold production process. In order to enable the introduction of spheroids with diameters more than 65 μm in height on an mLSI chip platform, a new mold fabrication method for round flow channels was required.We replaced the standard photolithography production process of the PDMS silicon molds by high precision milling. Upon milling with a cone-shaped milling tool, negative plastic PDMS prototyping molds with semi-circular microchannel profiles can be created with an accuracy of ±4 μm and surface roughness of ±1 μm. Initially, we milled four simple positive PMMA molds with single straight flow channels with a height of 100, 225, 360 and 500 μm and aspect ratios of 1. To test whether the flow channels could be closed by pneumatic membrane valves, the positive PMMA mold was inverted to a negative PDMS mold. Additionally, a corresponding control mold with a single microchannel for a pneumatic membrane valve in push-up configuration was manufactured using standard photolithography. The width of the valve elements on the silicon mold equaled the width of the corresponding flow channels, whereas the height was kept constant at 80 μm. Then, a two-layered PDMS chip for each different flow channel height was manufactured. All flow channels were successfully closed using the pneumatic membrane valve. The PDMS membrane thickness required was determined to be at least 25 μm. Below this thickness, a membrane would tear during actuation. A representative closing result of a pneumatic membrane valve with a height of 360 μm is shown in Fig. 1. A minor increase in closing pressure (from 18 to 21 psi) for the four flow channels at a thickness of 25 μm was observed with increasing channel height. The maximum closable channel height in a two-layered PDMS chip is determined using the highest possible spinning height of PDMS for formation of the deflective membrane. The minimum spin speed for obtaining reproducible PDMS films was found to be ∼250 rpm, which corresponds to a PDMS layer thickness of ∼750 μm.35 Subtracting the 25 μm required for the deflective PDMS membrane allows a maximum microchannel height of 725 μm that can be closed on mLSI chip platforms by using milled molds.
 | ||
| Fig. 1 The macro valve. Bright-field images of pneumatic membrane valve with a dimension for the cross section between the control and flow channel of 360 μm. The lower image shows a microchannel with a semi-circular round profile obtained by PDMS prototyping from milled PMMA molds. | ||
The mClear-Chip
The mClear-Chip is an mLSI chip platform consisting of four layers, including two thin polydimethylsiloxane (PDMS) layers bonded to a 4 mm PMMA top plate and a 0.17 mm glass bottom substrate (see Fig. 2A). The layout of the PMMA top plate is shown in Fig. 2B. A stiff material for the top layer of the chip was used to standardize reservoirs for chemicals, pressure connection lines, and inlet ports for biological material. The reagent reservoirs within the PMMA layer each can hold up to 28 μl. To minimize the pressure connection to the chip, eight reagent reservoirs are combined to single addressable pressure compartments for the flow channels, where the pressure is applied by removable and reusable PMMA adaptors (see Fig. 2C). The pressure adaptors and the top chip layer are tightly connected with four screws and individually sealed with O-rings. Pressure connections to close and open individual pneumatic membrane valves in the lower PDMS layers are connected to an additional PMMA adaptor, in which O-rings seal the individual ports.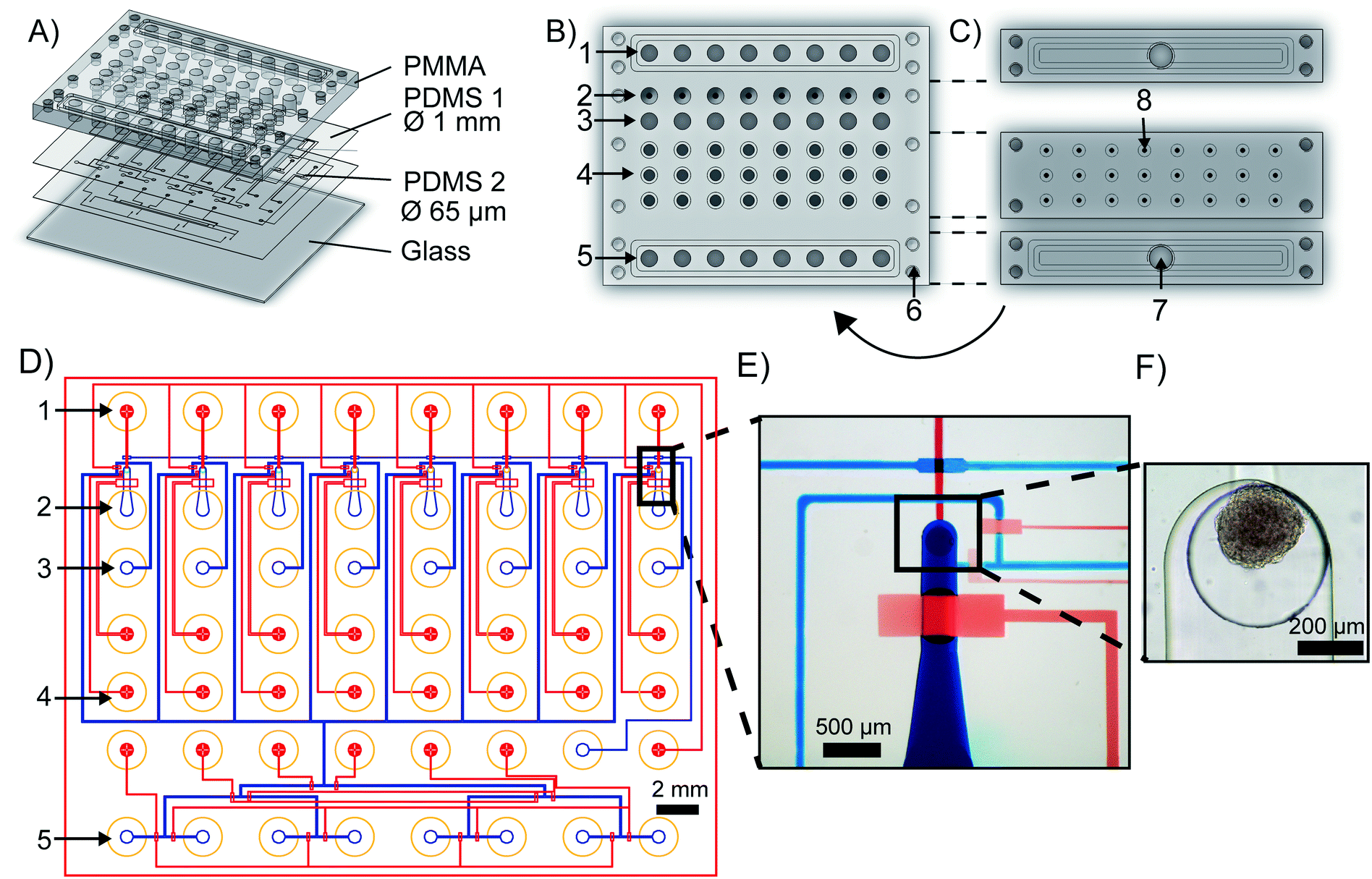 | ||
| Fig. 2 The mClear-Chip. A) The four different layers of the microfluidic large-scale integration chip platform for spheroid clearing. B) and C) The PMMA top layer with reagent reservoirs and standardized pressure adaptors. 1 – Fluid outlet reservoirs, 2 – spheroid inlet, 3 – fluid outlet, 4 – water reservoirs for pneumatic membrane valves, 5 – fluid inlet reservoirs, 6 – screw holes for mounting top plate, 7 – fluid pressure inlet port, 8 – pressure inlet ports for the mLSI valves. D) Images of the top (blue) and lower (red) PDMS layer. Yellow circles denote the inlet ports within the PMMA layer. E) Image of the mLSI chip unit cell. F) Trapped spheroids in a microfluidic unit cell. | ||
The fluidic channel network of the two thin PDMS layers is shown in Fig. 2D, where the microchannel of the upper PDMS layer are stained blue and the microchannels of the lower layer are red. Both PDMS layers integrate fluidic and control microchannels. Pneumatic membrane valves in the top and bottom PDMS layers work in a push-down and push-up configuration, respectively. To separate the functions of the chip, the layout can be divided into one reagent inlet multiplexer and eight spheroid-processing unit cells. A representative close-up of one spheroid processing unit is shown in Fig. 2E, and a trapped spheroid on that same unit is highlighted in Fig. 2F. Spheroids are introduced through the main support channel (520 × 250 μm, width × height). Upon flushing the spheroid toward the outlet channel, it reaches a cylindrical via structure (400 × 320 μm, width × height), which interconnects the fluidic channel networks of the upper and lower PDMS layer. The via acts as an spheroid-trapping structure since the outlet channel connected to the via has a height of only 20 μm. Back flow of the spheroid to the spheroid inlet channel is restricted by closing a macro valve. The fabrication process of the mClear-Chip with a sequential layer pick-up method is given in detail within the material and method section.
Integration of CLARITY on chip
All experiments were performed with spheroids formed from hASCs. One spheroid was introduced into each of the eight spheroid inlet reservoirs of the chip by pipetting. On average, the diameters of the hASC spheroids were 200 ± 50 μm. The PMMA pressure adaptor plates were connected to the top plate. In order to make the spheroid flow into the via trapping structure, a negative flow pressure of 2 psi was applied at the outlet. Then, the macro valve was closed and the microenvironment of the spheroid was controlled by routing fluids from the fluid inlet reservoirs, through the trapping chamber towards the fluid outlet reservoirs of the mClear-Chip. The CLARITY process (Fig. 3A–D) was initiated with fixation of the spheroids by using a solution containing paraformaldehyde, acrylamide, and bisacrylamide at room temperature. Spheroids were soaked in the fixation solution for 1 h, with replacement of the fixation solution every 10 min. To achieve spatial polymerization of the hydrogel in only the spheroid tissue rather than in the entire microfluidic channel, we rapidly exchanged the fixation solution containing the hydrogel monomers with the thermal radical initiator in less than 2 s, while concomitantly raising the chip temperature to 37 °C. The rapid temperature shift in the chip was achieved using a fast-reacting thermal ITO heating plate. During the hydrogel polymerization step, biomolecules containing an amino group are crosslinked to the hydrogel mesh. After 2 h of polymerization, the thermal radical initiator/starter solution was exchanged with PBS. To extract the lipid content from the hydrogel-tissue mesh, a clearing solution containing 140 mM sodium dodecyl sulfate (SDS) was introduced into the chambers containing the spheroids. The chip was programmed to exchange the clearing solution every 30 min. After 3 days, the spheroid was passively cleared. An hASC spheroid immediately after the hydrogel fixation within the chip, and the same spheroid after 3 days of clearing is shown in Fig. 3E. | ||
| Fig. 3 Process overview for optical clearing of spheroids on a microfluidic large-scale integration chip platform. Individual steps with A) spheroid loading, B) spheroids soaking with the hydrogel solution, C) spatial hydrogel-tissue polymerization, and D) passive lipid extraction by repetitive perfusion of the clearing solution. E) Bright-field and corresponding fluorescence images of an hASC spheroid successfully cleared on the chip platform. Scale bars denote for 125 μm. | ||
For comparison of the on-chip tissue-clearing procedure applied on spheroids, we additionally cleared hASC spheroids off-chip in a 96-well plate. The off-chip clearing experiment was conducted to simulate the on-chip method with the difference of 100 μl clearing solution per well was exchanged every day. An equal transparency of the spheroids for the off-chip cleared spheroids was not observed until 12 to 14 days. The reduced clearing time for the on-chip method can be explained by the constant removal of the clearing solution. Despite this time advantage for the spheroid clearing process, a three-day clearing time is still too long to comply with higher throughput studies.
The original clearing performed on a mouse brain20 was accelerated by applying an electrical field gradient to the tissue. At a given concentration, the negatively charged SDS molecules form micelles with a high electrophoretic mobility. Lipids from tissue samples are extracted by partitioning into the SDS vesicles. Thus, increasing the mobility of the SDS vesicles through the tissue leads directly to a decreased lipid clearing time. However, integration of electrodes complicates the chip production process. Alternatively, we observed that the size of the hydrogel embedded-spheroid rapidly changed depending on the buffer conditions. We utilized this previously reported phenomenon to decrease the long clearing process.36
Osmotic pumping for lipids extraction from hydrogel-embedded spheroids
All hydrogels have a swelling capacity. Hydrogel swelling causes an increase in volume by uptake of water. On a molecular level, water in a hydrogel is either bonded to polar hydrophilic groups or fills the space between the pores or voids, which is described as free-moving water. The swelling properties of the hydrogel primarily depend on the hydrogel crosslinking degree, solvent/polymer interaction, and ionic charge of the hydrogel polymer. The hydrogel formed during the CLARITY procedure is a heteromeric hydrogel composed of polyacrylamide and cellular proteins and nucleic acids. Both biomolecule classes contain side chains, which can protonate and deprotonate. Consequently, the neutral polyacrylamide hydrogel turns into a charged hydrogel. Lipid clearing of the tissue-embedded hydrogel occurs at pH 8.5, which is reportedly accompanied with significant hydrogel swelling. Upon exchange of the buffer back to pH 7.2, the hydrogel-embedded tissue shrinks. We confirmed this behavior by measuring the swelling and shrinkage kinetics of a hydrogel-embedded hASC spheroid upon a fast and reversible exchange of the pH within the trapping chamber of the chip. The changing spheroid's volume in response to the pH change from 5.5 (MES buffer) to 8.5 (boric acid buffer) is shown in Fig. 4A. Notably, the osmolality of both buffers was kept constant. Average spheroid diameters (n = 16) were obtained by analysis of time-lapse acquired bright-field images, whereas the spheroid volume was calculated by assuming a symmetric spheroidal structure. The first order rate constant for swelling and shrinkage of the hydrogel-embedded spheroid upon inducing the pH jump was 0.36 and −0.12 min−1, respectively.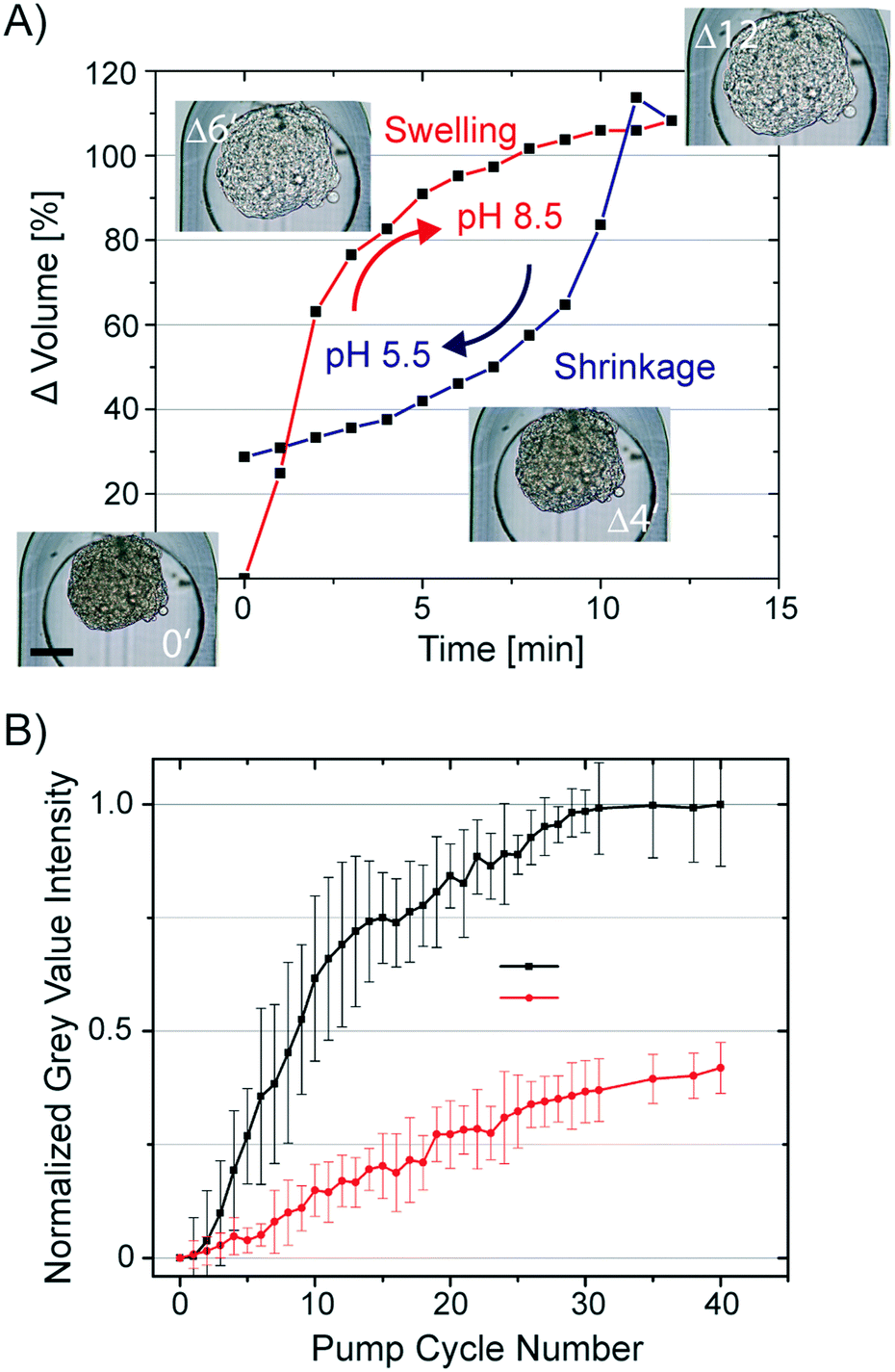 | ||
| Fig. 4 Osmotic pumping on the chip to decrease the spheroid clearing time. A) Kinetics of the spheroid volume changes in dependence of the surrounding pH value on the chip. Insets showing representative images of a hydrogel-embedded hASC spheroid cleared on the chip at the different pH values and response times upon pH change. B) The black and red graph shows the increased grey value intensity of spheroids subjected to repetitive osmotic pump cycles and perfusion with the clearing solution, respectively. In each osmotic pump cycle, the clearing solution (pH 8.5) was exchanged with a PBS buffer (pH 6.5) in intervals of 5 min. In the control experiment, only the clearing solution was used. | ||
Swelling and shrinkage of charged hydrogels is well documented in the literature and explained by osmosis.37 The water movement towards the hydrogel at higher pH can be explained by the gain of positive charges due to de-protonation of amino acids crosslinked to the hydrogel mesh. To equalize the osmolality of the buffer, water is directed to the hydrogel and leads to swelling. The opposing protonation of the amino acids in the hydrogel at lower pH can account for the shrinkage of the hydrogel.
To utilize the swelling/shrinking property of the tissue-embedded hydrogel to decrease the clearing time of the tissue, we repetitively subjected hydrogel-embedded, but non-cleared, spheroids to pH changes. It was predicted that lipid extraction could be facilitated by osmotic pumping. We exchanged the clearing solution (pH 8.5) with a phosphate buffer (pH 6.5) and back in 10 min intervals. The volume changes of spheroids induced by lowering the pH value was 55 ± 5%. To determine the clearing efficiency of osmotic pumping, bright-field images of the spheroids were taken every min over 6.5 h. Images were analyzed by segmentation of the spheroids and calculation of average grey value intensity. All grey value intensities were normalized to the value measured at the end-point of the clearing process of the individual spheroid. In Fig. 4B, the normalized grey value intensity of the spheroids is plotted against the osmotic pump cycle. For comparison, we repeated the experiment without pH shifts by only refreshing the clearing solution around the spheroids in the same time interval as in the osmotic pumping approach. The transparency values of the spheroids obtained from the image analysis are listed in Fig. 4B. It is apparent that after 30 osmotic pump cycles, the optical transparency of spheroids showed no additional marginal improvement. The grey value intensity of spheroids perfused with clearing solution increased only to 0.45 as compared to the spheroids subjected to 30 osmotic pump cycles. Furthermore, the grey value intensities for the spheroids cleared by refreshing the clearing solution every 5 min increased to the maximum after 2 days. Conclusively, the osmotic pumping significantly accelerated the clearing process from 2 days to 5 h (a factor of 10).
Fluorescence imaging of cleared spheroids on chip
To demonstrate that the osmotic pump processes does not lead to a disruption of the relative spatial cellular organization, we acquired fluorescence images of the DNA and protein content in spheroids. For this, cleared spheroids with the osmotic pump method were perfused on the chip with DAPI to stain the nuclear structure. Fig. 5A shows a color coded maximum intensity projection of an entire cleared spheroid with a diameter of 150 μm. The right panel of Fig. 5A shows that the DAPI fluorescence intensity did not decrease upon imaging through the entire spheroid. Although initial sizes of investigated spheroid were ∼200 μm, upon introducing CLARITY mounting media to match the refractive index average diameter shrink to 150 μm. The image shows that the functionality of the chip integrated CLARITY method. Furthermore, it is visible that the osmotic pump cycles did not perturb the nuclear structure. To proof that the proteins maintained their positions within the cleared spheroids we further, perfused the cleared spheroids on the same chip with fluorescently labeled anti-COX IV and anti-GAPDH to stain the mitochondria and cytoplasm, respectively. Spheroids were incubated for 12 h with the antibody solutions. The antibody solution was exchanged in the spheroid chamber on chip every 30 minutes. The mitochondrial protein COX-IV and GADH images are shown in Fig. 5B and SI2,† respectively. Both proteins could be detected with equal fluorescence intensity within the entire spheroid. For both labeled antibodies, we observed that the fluorescence intensity decreased with the imaging depth but it was still possible to image through the entire spheroids.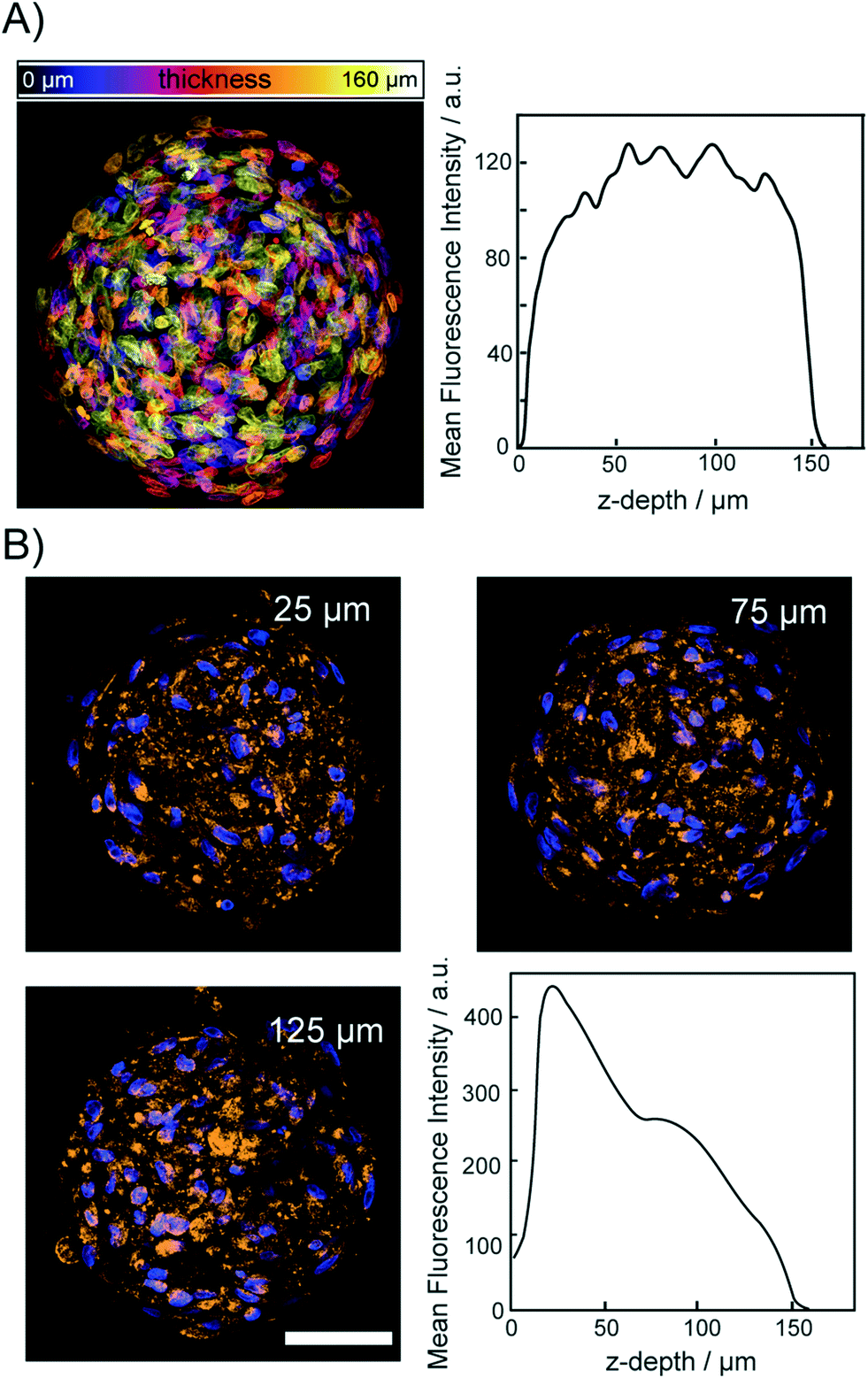 | ||
| Fig. 5 Fluorescence images of cleared spheroids. All spheroids were fixed following the CLARITY approach and cleared by osmotic pumping described in the main text. A) Maximum intensity projection of DAPI stained hASC spheroid. B) Representative fluorescence image sections through hASC spheroids stained with anti-COX-IV (Alexa647). Right lower panel shows the average fluorescence intensity of all image sections of the spheroid in dependence of the imaging depth. While the brightness of the images in B were gradually adjusted with the z-depth, the laser power during acquisition was not. | ||
Discussion
Microfluidic large-scale integration technology has proven to be an enabling technology for high-throughput 2D cell cultures.23,38 The strength of chip technologies lies in the automation and dynamic control of the cellular microenvironment of cell cultures over long time periods, which is required for stem cell differentiation.39 In particular, the possibility to integrate complex analytical workflows on the same platform for in situ protein or RNA detection makes the mLSI technology attractive, despite the complex controller hardware.40 In this study, we developed a production process based on plastic rapid prototyping molds for mLSI chips, which provides (i) a standardized pressure connector interface that can be applied in research to reduce the hardware footprint, and (ii) a pneumatic “macro valve,” which allows for the introduction of any type of biological sample with a diameter up to 700 μm, including biopsies or organoids, on an mLSI chip. The new mLSI manufacturing process allowed us to develop mClear-Chip for automatization of 3D cell culture clearing.The functionality of mClear-Chip was demonstrated by clearing the spheroids with a fully integrated CLARITY protocol. Current tissue-clearing protocols are based on fixation chemistry, hydrogel compositions, and single-molecule detection.41,42 Therefore, multiplexing spheroid cultures under standardized conditions is highly desirable for the development of new clearing protocols. Although the advances of mLSI technology for tissue clearing were demonstrated for processing eight spheroids in parallel, the modular unit cell design allows for rapid upscaling to higher sample numbers. The described microfluidic flush protocol with six alternating solutions in 30 flush operations for the CLARITY procedure is also applicable to any microfluidic chip platform; however, to the best of our knowledge, it can only be fully automated with PMV logics using mLSI technology. Furthermore, the standard interface allows for retrieving the samples for other downstream analyses such as mass spectrometry or next-generation sequencing. Automation of the CLARITY procedure in combination with the osmotic pumping approach led to a decrease in the clearing time for spheroids by a factor of approximately 50 compared to passive spheroid clearing off-chip. This is equivalent to the reduction achieved by applying an electric field gradient to a tissue sample, but avoids the risk of damage to the tissue caused by the high-strength electric field.
Deep-tissue imaging using the CLARITY approach on closed microfluidic chip platforms has the drawback that the glass chip substrate on the bottom and the PDMS chip on the top could hinder direct contact of the objective with the spheroid sample. Thus, a refractive index mismatch between the objective and the sample cannot be avoided. Concomitantly, this can explain the observe decrease in the fluorescence intensity of the antibodies in deeper imaging layers of the spheroids.
In contrast to single-plane illumination microscopy techniques combined with microfluidics,18,43 imaging the cleared spheroids is simple, and results in comparable resolution. The disadvantage of any tissue-clearing method is that this is an endpoint detection method. Thus, in order to obtain temporal resolution of biological processes, time trajectories have to be prepared with multiple specimens. This will be possible upon upscaling of our unit cells.
A recent study using expansion microscopy demonstrated that the relative spatial order of cross-linked biomolecules is maintained in a hydrogel-embedded tissue.21 In the future, the mClear-Chip can be applied to study biomolecular signals during long-term stem cell differentiation in 3D tissue models, and resolving signal transduction between cells. This will be particularly helpful for the maturation of adipocytes from stem cells, since they possess a high lipid content; thus, the gained resolution will improve analysis of mechanical, chemical, and cyto-architectural signals govern their maturation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the German Excellence Initiative of the German Federal and State Governments (EXC-294) and by the German Research Foundation (Emmy-Noether Grant ME3823/1-1). We thank the staff of the Life Imaging Center (LIC) in the Center for Biological Systems Analysis (ZBSA) of the Albert-Ludwigs-University Freiburg for help with their confocal microscopy resources, and the excellent support in image recording and analysis. We want explicitly thank A. Naumann, and R. Nitschke.References
- A. Fatehullah, S. H. Tan and N. Barker, Nat. Cell Biol., 2016, 18, 246–254 CrossRef PubMed.
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS PubMed.
- H. Clevers, Cell, 2016, 165, 1586–1597 CrossRef CAS PubMed.
- W.-Y. Lu, T. G. Bird, L. Boulter, A. Tsuchiya, A. M. Cole, T. Hay, R. V. Guest, D. Wojtacha, T. Y. Man, A. Mackinnon, R. A. Ridgway, T. Kendall, M. J. Williams, T. Jamieson, A. Raven, D. C. Hay, J. P. Iredale, A. R. Clarke, O. J. Sansom and S. J. Forbes, Nat. Cell Biol., 2015, 17, 971–983 CrossRef CAS PubMed.
- A. Petrova, A. Celli, L. Jacquet, D. Dafou, D. Crumrine, M. Hupe, M. Arno, C. Hobbs, A. Cvoro, P. Karagiannis, L. Devito, R. Sun, L. C. Adame, R. Vaughan, J. A. McGrath, T. M. Mauro and D. Ilic, Stem Cell Rep., 2014, 2, 675–689 CrossRef CAS PubMed.
- E. Fennema, N. Rivron, J. Rouwkema, C. van Blitterswijk and J. de Boer, Trends Biotechnol., 2013, 31, 108–115 CrossRef CAS PubMed.
- E. W. K. Young and D. J. Beebe, Chem. Soc. Rev., 2010, 39, 1036–1048 RSC.
- Y.-C. Tung, A. Y. Hsiao, S. G. Allen, Y.-S. Torisawa, M. Ho and S. Takayama, Analyst, 2011, 136, 473–478 RSC.
- J. Landry, D. Bernier, C. Ouellet, R. Goyette and N. Marceau, J. Cell Biol., 1985, 101, 914–923 CrossRef CAS PubMed.
- V. I. Khaoustov, G. J. Darlington, H. E. Soriano, B. Krishnan, D. Risin, N. R. Pellis and B. Yoffe, In Vitro Cell. Dev. Biol.: Anim., 1999, 35, 501–509 CrossRef CAS PubMed.
- B. Patra, Y.-H. Chen, C.-C. Peng, S.-C. Lin, C.-H. Lee and Y.-C. Tung, Biomicrofluidics, 2013, 7, 054114 CrossRef PubMed.
- W. Liu, J.-C. Wang and J. Wang, Lab Chip, 2015, 15, 1195–1204 RSC.
- A. Y. Hsiao, Y.-S. Torisawa, Y.-C. Tung, S. Sud, R. S. Taichman, K. J. Pienta and S. Takayama, Biomaterials, 2009, 30, 3020–3027 CrossRef CAS PubMed.
- V. van Duinen, S. J. Trietsch, J. Joore, P. Vulto and T. Hankemeier, Curr. Opin. Biotechnol., 2015, 35, 118–126 CrossRef CAS PubMed.
- S. H. Au, M. D. Chamberlain, S. Mahesh, M. V. Sefton and A. R. Wheeler, Lab Chip, 2014, 14, 3290–3299 RSC.
- A. D. Gracz, I. A. Williamson, K. C. Roche, M. J. Johnston, F. Wang, Y. Wang, P. J. Attayek, J. Balowski, X. F. Liu, R. J. Laurenza, L. T. Gaynor, C. E. Sims, J. A. Galanko, L. Li, N. L. Allbritton and S. T. Magness, Nat. Cell Biol., 2015, 17, 340–349 CrossRef CAS PubMed.
- R. P. J. Barretto and M. J. Schnitzer, Cold Spring Harb. Protoc., 2012 DOI:10.1101/pdb.prot071472.
- F. Pampaloni, N. Ansari and E. H. K. Stelzer, Cell Tissue Res., 2013, 352, 161–177 CrossRef PubMed.
- D. Zhu, K. V. Larin, Q. Luo and V. V. Tuchin, Laser Photonics Rev., 2013, 7, 732–757 CrossRef CAS PubMed.
- K. Chung and K. Deisseroth, Nat. Methods, 2013, 10, 508–513 CrossRef CAS PubMed.
- F. Chen, P. W. Tillberg and E. S. Boyden, Science, 2015, 347, 543–548 CrossRef CAS PubMed.
- P. M. Fordyce, C. A. Diaz-Botia, J. L. DeRisi and R. Gómez-Sjöberg, Lab Chip, 2012, 12, 4287 RSC.
- R. Gómez-Sjöberg, A. Leyrat, D. Pirone, C. Chen and S. Quake, Anal. Chem., 2007, 79, 8557 CrossRef PubMed.
- M. Blazek, T. S. Santisteban, R. Zengerle and M. Meier, Lab Chip, 2015, 15, 726–734 RSC.
- J. M. Yuhas, A. P. Li, A. O. Martinez and A. J. Ladman, Cancer Res., 1977, 37, 3639–3643 CAS.
- M. Meier, R. V. Sit and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 477–482 CrossRef CAS PubMed.
- A. Tropmann, L. Tanguy, P. Koltay, R. Zengerle and L. Riegger, Langmuir, 2012, 28, 8292–8295 CrossRef CAS PubMed.
- L. Gitlin, P. Schulze and D. Belder, Lab Chip, 2009, 9, 3000–3002 RSC.
- T. S. Santisteban, R. Zengerle and M. Meier, RSC Adv., 2014, 4, 48012–48016 RSC.
- K. Kim, S. W. Park and S. S. Yang, BioChip J., 2010, 4, 148–154 CrossRef CAS.
- J. White and A. M. Streets, bioRxiv, 2017, pp. 1–18 Search PubMed.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS PubMed.
- V. Studer, G. Hang, A. Pandolfi, M. Ortiz, W. French Anderson and S. R. Quake, J. Appl. Phys., 2004, 95, 393–398 CrossRef CAS.
- E. P. Kartalov, A. Scherer, S. R. Quake, C. R. Taylor and W. F. Anderson, J. Appl. Phys., 2007, 101, 064505 CrossRef PubMed.
- W. Y. Zhang, G. S. Ferguson and S. Tatic-Lucic, Elastomer-supported cold welding for room temperature wafer-level bonding, IEEE, 2004, pp. 741–744 Search PubMed.
- K. Chung, J. Wallace, S.-Y. Kim, S. Kalyanasundaram, A. S. Andalman, T. J. Davidson, J. J. Mirzabekov, K. A. Zalocusky, J. Mattis, A. K. Denisin, S. Pak, H. Bernstein, C. Ramakrishnan, L. Grosenick, V. Gradinaru and K. Deisseroth, Nature, 2013, 497, 332–337 CrossRef CAS PubMed.
- H. H. Hooper, J. P. Baker, H. W. Blanch and J. M. Prausnitz, Macromolecules, 1990, 23, 1096–1104 CrossRef CAS.
- M. Blazek, C. Betz, M. N. Hall, M. Reth, R. Zengerle and M. Meier, Mol. Cell. Proteomics, 2013, 12, 3898–3907 CAS.
- G. G. Giobbe, F. Michielin, C. Luni, S. Giulitti, S. Martewicz, S. Dupont, A. Floreani and N. Elvassore, Nat. Methods, 2015, 12, 637–640 CrossRef CAS PubMed.
- X. Wu, N. Schneider, A. Platen, I. Mitra, M. Blazek, R. Zengerle, R. Schüle and M. Meier, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4143–E4150 CrossRef CAS PubMed.
- M.-T. Ke, S. Fujimoto and T. Imai, Nat. Neurosci., 2013, 16, 1154–1161 CrossRef CAS PubMed.
- H. Hama, H. Kurokawa, H. Kawano, R. Ando, T. Shimogori, H. Noda, K. Fukami, A. Sakaue-Sawano and A. Miyawaki, Nat. Neurosci., 2011, 14, 1481–1488 CrossRef CAS PubMed.
- P. Paiè, F. Bragheri, A. Bassi and R. Osellame, Lab Chip, 2017, 16, 1556–1560 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7lc01114h |
| This journal is © The Royal Society of Chemistry 2018 |