
Formulation and catalytic performance of MOF-derived Fe@C/Al composites for high temperature Fischer–Tropsch synthesis†
Lide
Oar-Arteta
a,
María José
Valero-Romero
a,
Tim
Wezendonk
a,
Freek
Kapteijn
 a and
Jorge
Gascon
*ab
a and
Jorge
Gascon
*ab
aFaculty of Applied Sciences, Chemical Engineering, Delft University of Technology, Building 58, Van der Maasweg 9 2629HZ, Delft, The Netherlands
bKing Abdullah University of Science and Technology, KAUST Catalysis Center, Advanced Catalytic Materials, Thuwal 23955, Saudi Arabia. E-mail: jorge.gascon@kaust.edu.sa
First published on 15th November 2017
Abstract
High productivity towards C2–C4 olefins together with high catalyst stability are key for optimum operation in high temperature Fischer–Tropsch synthesis (HT-FTS). Here, we report the fabrication of Fe@C/Al composites that combine both the outstanding catalytic properties of the Fe–BTC MOF-derived Fe catalyst and the excellent mechanical resistance and textural properties provided by the inorganic AlOOH binder. The addition of AlOOH to Fe–BTC followed by pyrolysis in N2 atmosphere at 500 °C results in composites with a large mesoporosity, a high Fe/Fe3O4 ratio, 10–35 nm average Fe crystallite size and coordinatively unsaturated Al3+ sites. In catalytic terms, the addition of AlOOH binder gives rise to enhanced C2–C4 selectivity and catalyst mechanical stability in HT-FTS, but at high Al contents the activity decreases. Altogether, the productivity of these Fe@C/Al composites is well above most known Fe catalysts for this process.
1. Introduction
Ethylene, propylene, and butylenes are essential building blocks in the chemical industry. These so-called lower or light olefins are among the organic chemicals with the largest production volumes worldwide.1 Their broad spectrum of derivatives results in a very diverse end market, ranging from packing materials and synthetic textiles to antifreezing agents, solvents, and coatings.2C2 to C4 range of olefins are traditionally produced through thermal or catalytic cracking of a range of hydrocarbon feedstocks, such as naphtha, gas oil, condensates and light alkanes.3,4 An interesting alternative is to produce lower olefins from synthesis gas, a mixture of CO and H2 derived from natural gas, coal or biomass. One of the processes that allows the conversion of synthesis gas into lower olefins without intermediate steps is the so called Fischer–Tropsch-to-olefins (FTO).5 This process is generally performed over iron-based catalysts at relatively high temperature to shift selectivity towards short-chain hydrocarbons. Iron catalysts have several advantages for olefin synthesis such as high selectivity, low methanation activity, high availability thus low price, and low sensitivity to poisons. Moreover, they are efficient catalysts for utilization of syngas produced from coal and biomass which have low hydrogen to carbon monoxide ratios (H2/CO < 2), typically around unity.6
Recently, metal organic frameworks (MOFs) have been applied as precursors for iron based catalysis, resulting in an unrivalled performance in high temperature Fischer Tropsch synthesis (HT-FTS) for the production of short chain hydrocarbons.7 In spite of high Fe loadings, the high dispersion of the active phase and its encapsulation in a highly porous carbon matrix give rise to very high activity and stability. As a result, the MOF-derived solids display productivities, on a total catalyst weight basis, one order of magnitude higher than that of the well-known Ruhrchemie and Sasol catalysts for HT-FTS.7 Moreover, when promoted with potassium, these catalysts show a higher activity due to an enhancement of the water gas shift functionality and a higher olefin to paraffin ratio due to modulation of the hydrogenation ability. This effect also leads to a significant decrease in methane selectivity.8
The relatively low mechanical strength constitutes however the main constraint towards industrial application of these materials. Despite their promising catalytic results, the MOF derived iron catalysts, after been shaped into pellets, display a suboptimal mechanical stability which may lead to segregation of the catalyst particles under HT-FTS conditions, which eventually leads to plugging of the catalytic bed in fixed-bed reactor operation.9
Inorganic binders such as clays are commonly added to porous powders to confer them the desired mechanical strength.10,11 Among them, boehmite (aluminum oxyhydroxide, AlOOH) is considered to be one of the most versatile colloidal binders.12 When boehmite is annealed, it undergoes a series of polymorphic phase transformations: from boehmite into γ-alumina and then into δ- and θ-alumina, known as transitional aluminas, and eventually into α-alumina, the thermodynamically most stable alumina phase.13
HT-FTS using Fe/γ-Al2O3 has been typically used to produce lower-olefins.14 Supported iron catalysts display enhanced dispersion of the active phase and may withstand the mechanical degradation during the HT-FTS process compared to the bulk iron catalysts. Nevertheless, research on supported iron catalysts has met with limited success, mainly due to their cumbersome activation. In this sense, highly dispersed iron oxide may interact strongly with the high surface area oxidic support, impeding the conversion of iron oxide into the active phase (iron carbide).15
Bringing needs and concepts together, this study focuses on the optimum formulation and testing of new particulate hybrid materials derived from MOFs with improved mechanical properties for HT-FTS. Here we present the properties of Fe@C/Al catalysts prepared from different mixtures of Fe–BTC (BTC: 1,3,5-benzenetricarboxylate) MOF and aluminium oxide hydroxide (AlOOH) as inorganic binder and their catalytic performance in HT-FTS in terms of catalytic activity, selectivity and catalytic and mechanical stability.
2. Experimental
2.1. Catalyst synthesis
The pure MOF derived Fe catalyst (denoted as Fe@C) was obtained by pyrolysis at 500 °C under continuous N2 flow (200 cmSTP3 min−1) of the pristine Fe–BTC MOF (Basolite F300, obtained from BASF). The pyrolysis temperature was reached at a heating rate of 5 °C min−1 and maintained for 4 h. The xFe@C/Al (x = iron content) composites were prepared by wet extrusion of different Fe–BTC (Basolite F300, BASF)/AlOOH (Disperal®, Sasol) mixtures with 90/10, 70/30 and 50/50 mass ratios and subsequently pyrolyzed under the same conditions. Before exposure to air, each material was passivated at room temperature using 5 vol% O2 in N2 for 2 h. Using 70/30 as standard mass ratio, two additional composites were prepared. The first material was pyrolyzed at lower temperature (400 °C) whereas the second material was obtained after wet extrusion of the pyrolyzed Fe–BTC at 500 °C with the fresh boehmite without further treatment. All the samples were then pelletized pelletized by applying 2 tonnes per cm2 pressure for 5 min, crushed and sieved to particle size range of 100–212 μm.2.2. Characterization of the materials
2.3. High temperature Fischer–Tropsch synthesis (HT-FTS) and preliminary bed integrity test under Fischer–Tropsch temperature and pressure conditions (FT-TP test)
HT-FTS experiments were performed in the six-flow fixed-bed microreactor (FBM) setup described elsewhere.20 The FTS setup allows running six experiments in parallel with identical temperature and composition of feed gas, whereas the flow rate of the feed and the pressure is tuneable for each individual reactor.Durability of the catalysts in a process is referred to as the time-on-stream (TOS) they are able to operate before segregation results in uncontrollable pressure drop over the reactor bed, thus it is directly related to the mechanical strength of the catalytic materials. In order to evaluate and compare the activity and stability of the pure 38Fe@C and the 33Fe@C/Al, 25Fe@C/Al and 15Fe@C/Al composites, the degree of dilution of the catalytic bed needed to be modified to avoid blockage of the reactors. Hence, variable mass from 50 to 150 mg of catalyst (100–212 μm) was loaded in the reactors in order to obtain the same amount of Fe and gas-hourly space velocity (GHSV) of 55 mmolCO,STP gFe−1 min−1 in all the reactors and mixed with SiC of similar size to keep a constant fixed bed volume of 1.3 cm3. First, samples were activated in 80 cmSTP3 min−1 of H2 at 400 °C for 3 h at atmospheric pressure followed by cooling down to 180 °C. After increasing the pressure to 15 bar, CO was gradually introduced in the feed stream at 180 °C until reaching a final H2/CO ratio of 1 and a total syngas flow of 50 cmsyngas,STP3 gFe−1 min−1. Finally, the reactor was heated to the reaction temperature of 340 °C. Permanent gases as well as light hydrocarbons in the gas phase were analyzed online by a Compact GC (Interscience) equipped with three columns and detectors in parallel using He as carrier gas. In the first column (Carboxen 1010, 10 m 0.32 mm) N2, CO, CH4 and CO2 were separated at 333 K and analyzed by TCD. In the second column (Al2O3/KCl, 10 m 0.32 mm) and FID detection, separation between all C1–C4 components was achieved at 434 K. In the third column (RTx-1 0.5 mm, 15 m 0.32 mm) C5–C10 hydrocarbons were separated at 353 K and analyzed by FID.
The catalytic activity is expressed in terms of both CO conversion (eqn (1)) and Fe time yield (FTY) (eqn (2)), defined as the number of moles of CO converted to hydrocarbon products, excluding CO2, per gram of Fe per second. The product selectivity (Si) (eqn (3)) and the yield of the C2–C4 hydrocarbon fraction (YC2–C4) were both determined after 50 h when a pseudo steady-state was reached and the first reactor loaded with a composite started blockage.
CO conversion (XCO), carbon selectivity (SCn) of each product and yield to olefins (YC2–C4=) have been calculated as follows (eqn (4)–(6)):
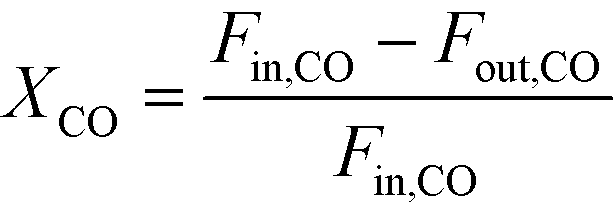 | (1) |
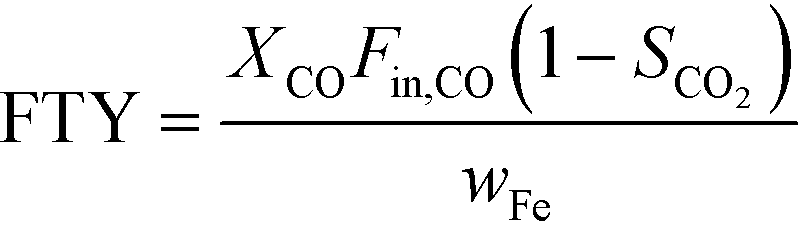 | (2) |
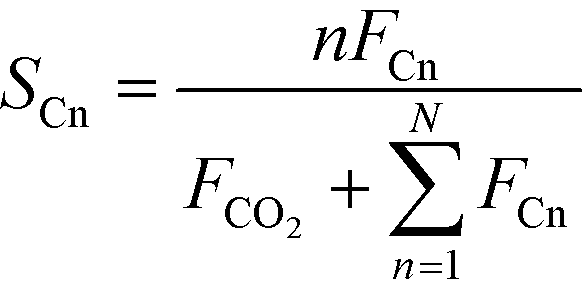 | (3) |
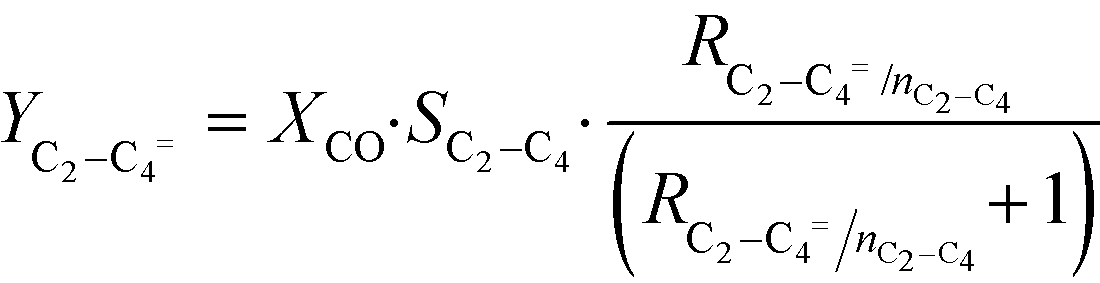 | (4) |
Here, F represents a molar flow rate. Besides, the α value is defined in terms of the rate of polymerization (rp) and the rate of termination (rt) of the growing hydrocarbons, according to eqn (5)
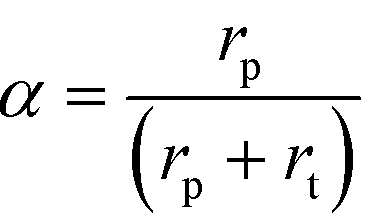 | (5) |
Eqn (6) represents the Anderson–Schulz Flory (ASF) product distribution in terms of molar fractions
| yCn = (1 − α)αn−1 | (6) |
Additionally, before testing the catalysts in HT-FTS, the materials were exposed to high temperature (340 °C) and pressure (15 bar) conditions typically used in HT-FTS under N2 flow (100 cm3 min−1) for a brief period of time (5 h) and their properties were analyzed before and after the test. This method is referred to as FT-PT onwards.
3. Results
3.1. Characterization
All materials have been thoroughly characterized before and after pyrolysis at 500 °C to understand the transformation taking place during the heat treatment in inert atmosphere, thus facilitating the interpretation of the catalytic performance in HT-FTS.During the pyrolysis at 500 °C the organic linker decomposes and partially volatilizes, being released in the form of carbonaceous products including free acids, anhydrides, carbon monoxide and carbon dioxide.21 Accordingly, an increase of the metal content is expected for all the MOF derived materials after the pyrolysis. Table S1† shows Fe loadings of the materials calculated from TGAs under air before and after the pyrolysis according to eqn (S1)–(S3).†
The iron loading of the carbonized materials decreases from 38 to 15 wt% when decreasing the Fe–BTC/AlOOH ratio from 100 to 50 due to the increasing presence of Al in the composites. As expected, after pyrolysis in N2, the increase in the iron loading is lower for the samples with lower Fe–BTC/AlOOH ratios. The increase in Fe loading varies from 42% to 33% for the pure Fe-BTC to 38FeAC and for the 90/10 Fe-BTC/AlOOH to 15FeAC/Al composite, respectively.
Besides metal loading, the metallic species together with their average crystal size play an essential role in the catalytic performance of the catalyst. Accordingly, the crystalline structure of the MOF, MOF-binder mixtures and the final Fe@C and Fe@C/Al composites have been analysed by PXRD. Fig. 1 shows diffraction patterns for Fe–BTC, boehmite and their composites before (Fig. 1a) and after (Fig. 1b) pyrolysis at 500 °C. The diffraction pattern of fresh Fe–BTC goes in line with reported data in literature for this Fe-based MOF.22 All detectable reflections in the pattern of boehmite can be assigned to orthorhombic γ-AlOOH with lattice parameters a = 3.699 Å, b = 12.22 Å, c = 2.86 Å and no additional reflections are observed when compared with the standard diffraction pattern (JCPDS File No. 21-1307) thus indicating the purity of the material.23,24 The signals of both diffraction patterns can be seen in the diffraction patterns of all fresh composite samples. The only difference among the composites is the MOF/oxide reflection ratio, confirming the different compositions of the samples. After pyrolysis, none of the materials show diffraction patterns of their precursors, evidencing the decomposition of the framework in the case of the MOF and full transformation of the AlOOH binder into γ-Al2O3. In situ measurements in our group have proven that the iron phases present in the 38Fe@C composite after pyrolysis at 500 °C consist of a mixture of metallic iron (Fe), iron(II) oxide (FeO) and iron carbides (FeC).8 The highly dispersed Fe particles are very pyrophoric after preparation, and the required passivation step at room temperature readily converts significant amounts of the Fe phase into Fe3O4.25 The ex situ XRD measurements carried out in this study show that cubic Fe3O4 is the predominant iron phase in 38Fe@C, however, a small diffraction peak at θ = 52° confirms the presence of metallic Fe. The signal of the diffraction peaks in the composites evolves according to their Fe@C/Al2O3 ratio. The sharp diffraction peak at θ = 52° in the 25Fe@C/Al and 15Fe@C/Al composites indicate the increasing crystal size of metallic Fe in these samples. The quantification of the crystalline species has been carried out with SIROQUANT® software by means of the Rietveld method.
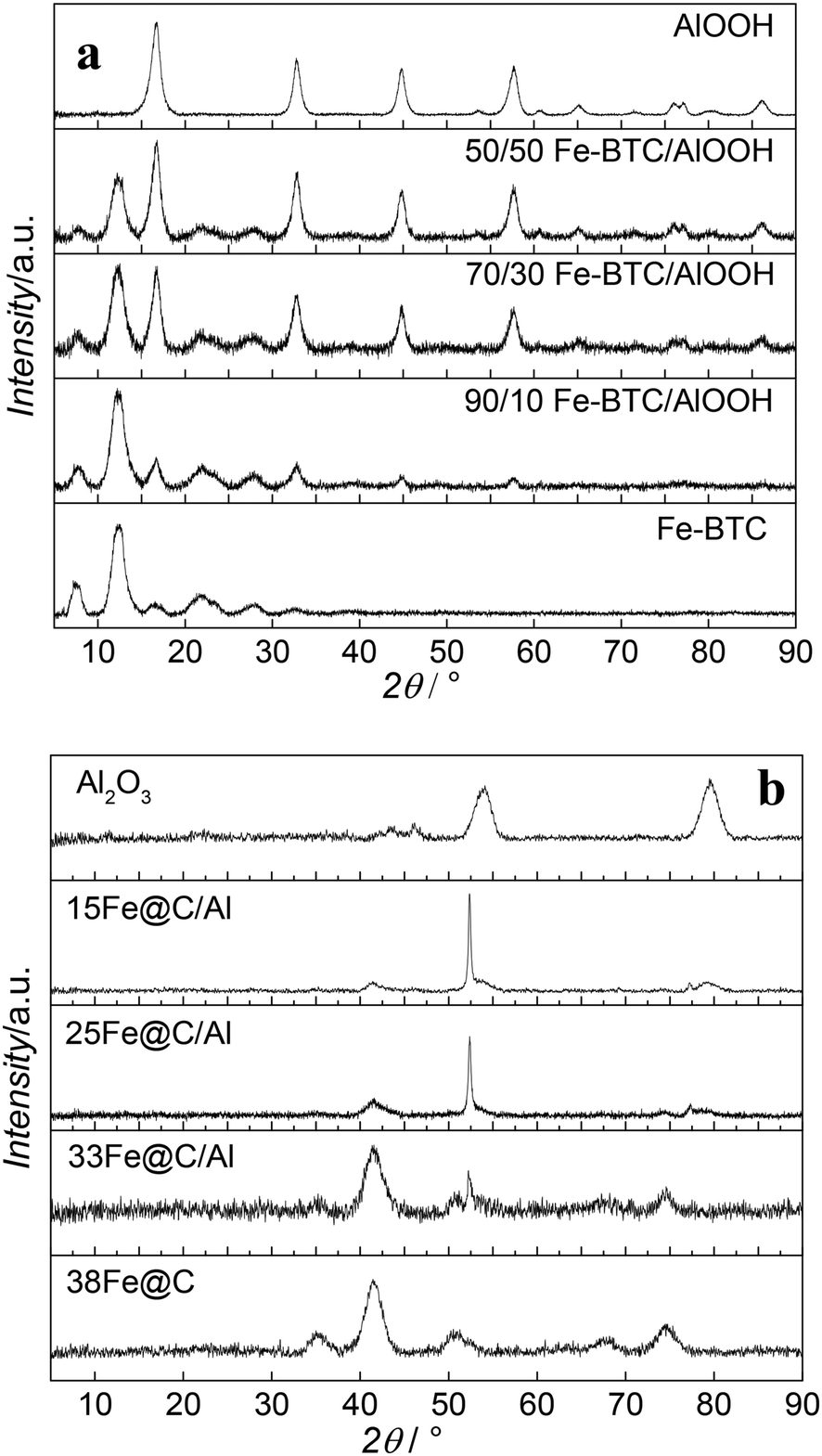 | ||
| Fig. 1 XRD patterns of Fe–BTC, boehmite and the composites before (a) and after (b) pyrolysis at 500 °C. | ||
Table S2† shows Fe/Fe3O4 ratios for the composites and the pure 38Fe@C and the mean crystallite sizes of metal and metal oxides of the pyrolyzed materials and after air exposure according to the Scherrer equation. Accordingly, the Fe/Fe3O4 ratio increases with increasing amount of binder as follows: 38Fe@C (0.07) < 33Fe@C/Al (0.26) < 25Fe@C/Al (2.63) < 15Fe@C/Al (5.93). Likewise, the Fe crystal size increases with the loading of binder in the catalyst from 3.8 nm for the pure 38Fe@C material to 36 nm for the 15Fe@C/Al composite. These results suggest that water released in the dehydration of AlOOH might contribute to the partial steam gasification of the carbon catalysed by Fe at 500 °C,26 thus favouring the sintering of Fe nanoparticles.
Textural properties of Fe–BTC and the boehmite before and after pyrolysis are shown in Table 1. Fe–BTC has very high surface area (SBET = 1010 cm2 g−1), but it drastically decreases after the heat treatment at 500 °C for 38Fe@C (SBET = 274 cm2 g−1). At the same time, the mesoporosity in 38Fe@C (Vmeso = 0.24 cm3 g−1) increases as a result of the decarboxylation of the trimesic acid linker during the pyrolysis of Fe–BTC (Vmeso = 0.06 cm3 g−1).21 Both boehmite (VTotal = Vmeso = 0.32 cm3 g−1) and boehmite-derived alumina (VTotal = Vmeso = 0.4 cm3 g−1) are purely mesoporous materials. The porosity of the latter, however, is higher due to the water released during the pyrolysis which partially blocks some of the pores in the fresh boehmite.27 Well in line with these results, the carbonized composites show intermediate surface areas of the materials that constitute them, namely, the carbonized Fe–BTC and the boehmite-derived alumina. Moreover, increasing the Al loading increases the total and mesoporous pore volume of the composites.
| Samples | S BET (cm2 g−1) | V meso (cm3 g−1) | V Total (cm3 g−1) |
|---|---|---|---|
| Fe–BTC | 1010 | 0.06 | 0.47 |
| AlOOH | 147 | 0.32 | 0.32 |
| 38Fe@C | 274 | 0.24 | 0.33 |
| Al2O3 | 132 | 0.4 | 0.4 |
| 33Fe@C/Al | 226 | 0.16 | 0.22 |
| 25Fe@C/Al | 160 | 0.26 | 0.28 |
| 15Fe@C/Al | 160 | 0.27 | 0.28 |
The DRIFT spectra of all fresh materials are shown in Fig. S1a and b† shows the DRIFT spectra for 38Fe@C, 25Fe@C/Al and Al2O3. The DRIFT spectra of pure boehmite and Fe–BTC in Fig. S1a† are consistent with those reported in literature.28 For the boehmite spectrum the main peaks are in the OH stretching region between 4000 and 2800 cm−1. The two intense and broad peaks at 3335 and 3108 cm−1 are due, respectively, to the asymmetric and symmetric stretching vibrations of bulk H-bonded hydroxyls which hold together the layers of boehmite.29 The shoulder at 3734 cm−1 is characteristic of the vibrations of the OH groups located at the lateral planes of boehmite.29,30 After pyrolysis, these diffraction peaks disappear (Fig. S1b†), evidencing the dehydroxylation of the boehmite at 500 °C. The spectrum of the Fe–BTC at room temperature contains vibrations of highly hydrated carboxylic acids. The broad absorbance around 2500–3600 cm−1 can be ascribed to the OH stretching of physisorbed water molecules in the pores of the MOF as well as contributions of OH stretching in the mentioned carboxylic acids.31,32 The small absorbance at 1890 cm−1 is a contribution of C–H bonds of aromatic compounds and the asymmetric νas(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and symmetric carboxylate stretchings νs(CO) are found around 1500–1750 cm−1 and 1300–1500 cm−1, respectively.33 The spectra of 38Fe@C and 25Fe@C/Al samples contain no large or sharp absorbances, which is caused by the removal of water from the compounds during the pyrolysis. The OH stretching and CH vibrations are absent from the spectra as well, due to framework decomposition during the pyrolysis. Altogether, the DRIFTS results evidence that i) the pre-pyrolysed MOF mixed boehmite looks exactly like a physical mixture of the two, as expected according to XRD (Fig. 1), ii) after pyrolysis, the characteristic AlOOH bands (2500–3500 cm−1) disappear and the spectra much resembles a dehydroxylized boehmite evolved towards Al2O3 and iii) the typical aromatic band contributions (asymmetric and symmetric CO stretch 1000–2000 cm−1) have disappeared as well, evidencing a carbonization of the organic linker toward non-functionalized pyrolytic and graphitic type carbon.
O) and symmetric carboxylate stretchings νs(CO) are found around 1500–1750 cm−1 and 1300–1500 cm−1, respectively.33 The spectra of 38Fe@C and 25Fe@C/Al samples contain no large or sharp absorbances, which is caused by the removal of water from the compounds during the pyrolysis. The OH stretching and CH vibrations are absent from the spectra as well, due to framework decomposition during the pyrolysis. Altogether, the DRIFTS results evidence that i) the pre-pyrolysed MOF mixed boehmite looks exactly like a physical mixture of the two, as expected according to XRD (Fig. 1), ii) after pyrolysis, the characteristic AlOOH bands (2500–3500 cm−1) disappear and the spectra much resembles a dehydroxylized boehmite evolved towards Al2O3 and iii) the typical aromatic band contributions (asymmetric and symmetric CO stretch 1000–2000 cm−1) have disappeared as well, evidencing a carbonization of the organic linker toward non-functionalized pyrolytic and graphitic type carbon.
TPR(H2) experiments have been conducted to study how the addition of the inorganic binder affects the reducibility of the metal oxide phase in the composites. To this purpose, both the consumption of H2 and the TPR profiles of the nanocomposites have been compared to that of the pure 38Fe@C catalyst and the results are shown in Fig. 2.
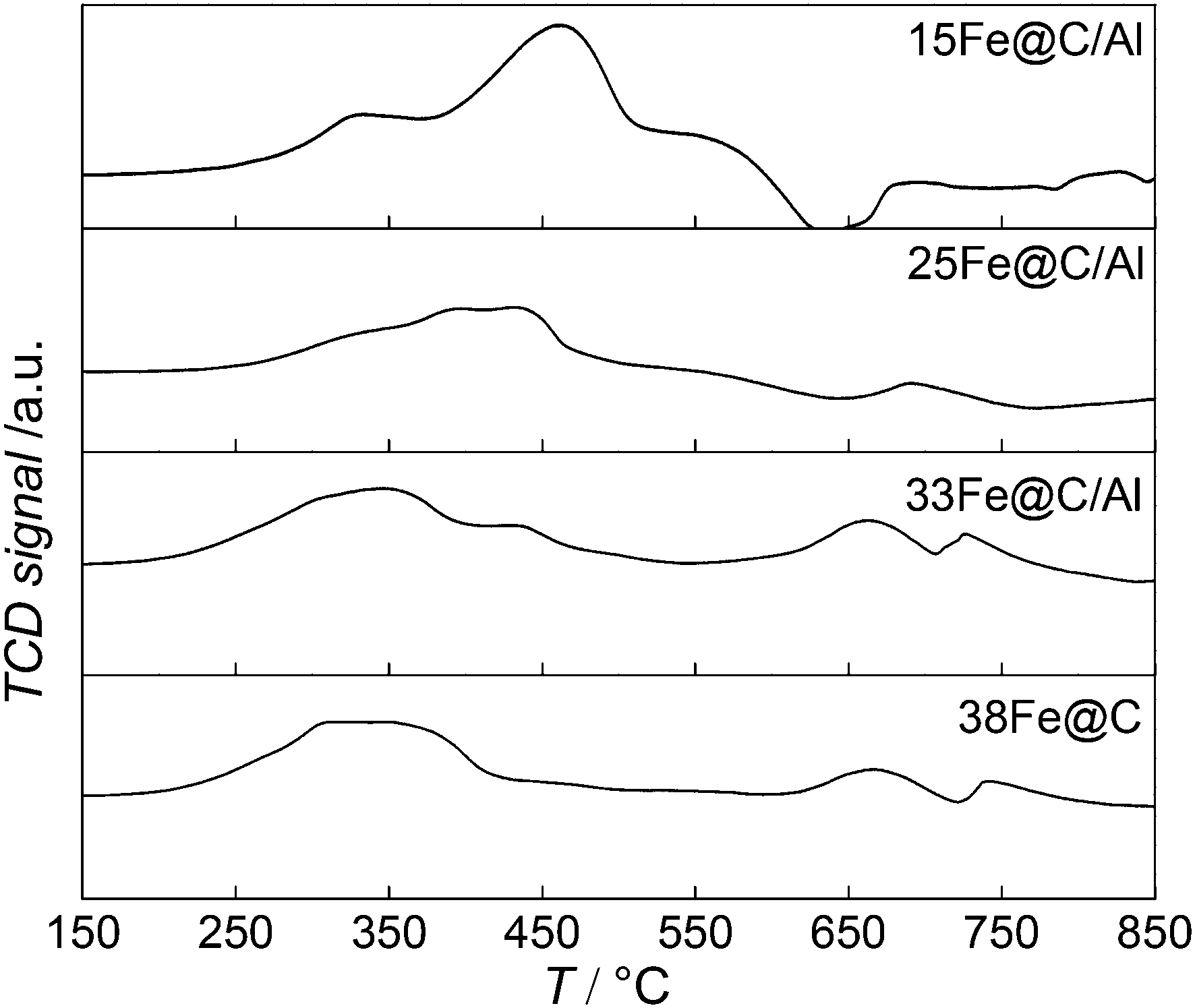 | ||
| Fig. 2 TPR profiles of 38Fe@C sample and the Fe@C/Al composites. | ||
All the samples exhibited two reduction steps, agreeing with the stepwise reduction of iron oxides as Fe3O4 → FeO → Fe.34 Thereby, the first reduction of Fe3O4 to FeO takes place followed by reduction to metallic Fe. In the case of 38Fe@C, both transformations take place in the same temperature range from 200 °C to 450 °C. For the composites the reduction processes shift towards higher temperature. Moreover, the higher the Al2O3 content in the composite the more pronounced the stepwise reduction takes place, thus highlighting the reduction of different iron species. As a matter of fact, the second step, i.e. the reduction of FeO to Fe, seems to be more hindered with increasing amount of Al2O3 than the first reduction step, i.e. the reduction of Fe3O4 to FeO. Previous work on the reducibility of iron oxides established that addition of H2O to the reducing gas affects especially the TPR peak for reduction of Fe3O4 to Fe metal, this peak shifting to higher temperatures and its shape changing considerably.35 This suggests that during the reduction of the composites, the amount of water released increases with the amount of binder thus shifting the reduction of Fe3O4 to higher T. In this manner, reduction peaks of the 15Fe@C/Al composite appear from 220 °C to 375° for Fe3O4 → FeO and from 380 °C to 500 °C for FeO/FeAl2O4 → Fe, thus shifting ∼50 °C to higher temperature compared to 38Fe@C. Altogether, these results point at a shift in the reduction process due to the release of water when increasing the amount of Al2O3 in the composite.
The acid sites of both pure AlOOH derived γ-Al2O3 and the 15Fe@C/Al have been studied by IR spectroscopy of adsorbed pyridine. Fig. 3 shows the IR spectra of adsorbed pyridine on both Al2O3 and the 15Fe@C/Al composite in the region of 1400–1700 cm−1.
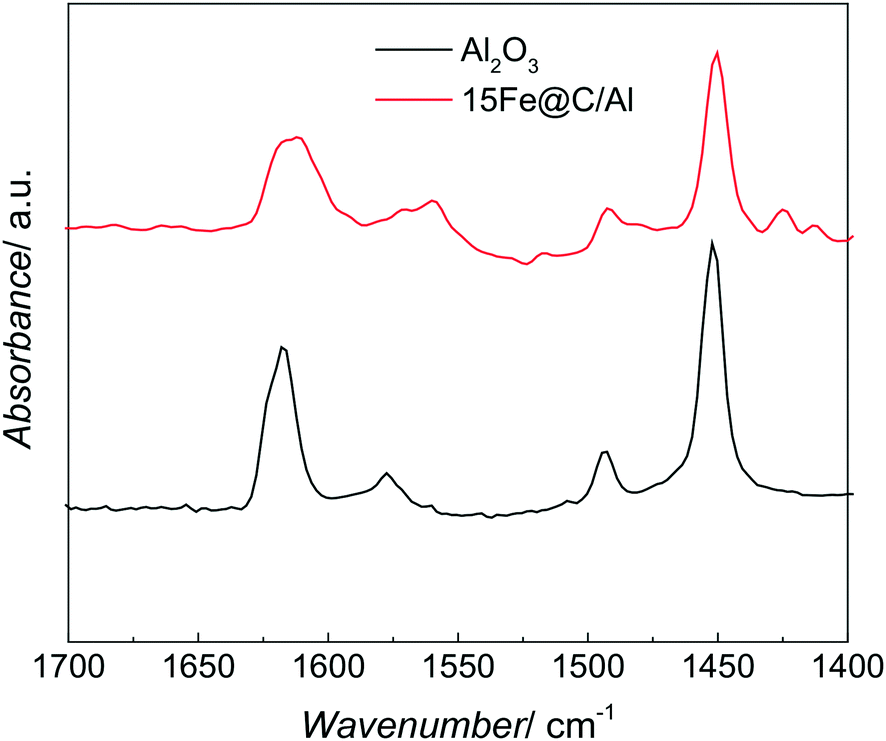 | ||
| Fig. 3 Representative IR spectra of pyridine adsorbed on the AlOOH derived Al2O3 and the 15Fe@C/Al composite. | ||
Both spectra show bands at 1450, 1490, 1598, 1612 and 1624 cm−1, which are characteristic of the ring vibration modes of adsorbed pyridine coordinating through the nitrogen lone pair to Lewis acid sites (most likely coordinatively unsaturated aluminium cations, Alcus3+).36 For the 38Fe@C and the 33Fe@C/Al, 25Fe@C/Al composites the amount of boehmite and thus the acid sites were non-existent or low, and thus could not be detected by this technique, although for the composites some Alcus3+ are expected.
SEM images show that 38Fe@C (Fig. 4a) has a disordered morphology with both plate like and spherical particles whereas 15Fe@C/Al (Fig. 4b) shows homogeneous granular morphology. SEM together with EDX scans of the 15Fe@C/Al (Fig. 4c) composite disclose how Fe (Fig. 4d) and Al (Fig. 4e) are homogeneously distributed in the sample. According to these images, this composite with the highest loading of binder consists of well dispersed Fe@C particles, evenly spread throughout the γ-Al2O3 matrix.
 | ||
| Fig. 4 SEM image of 38Fe@C (a) and 15Fe@C/Al (b). SEM picture (c) and EDX analysis of 15Fe@C/Al for Fe (d) and Al (e). | ||
Finally, TEM images (Fig. 5) show changes in the size of Fe nanoparticles and the shape of the 38Fe@C and Fe@C/Al composites. As the amount of binder precursor increases, the resulting material adopts the crystallite packing shape characteristic of mesoporous γ-Al2O3 from boehmite.37 The Fe PSD of 38Fe@C and the 33Fe@C/Al composite is shown in Fig. S2.† The 38Fe@C (Fig. S2a†) sample shows a narrow particle size distribution (PSD) with an average particle size between 2.5 and 8.0 nm. The width of the PSD increases considerably when adding binder to the sample and significantly shifts toward larger particle sizes. Hence, for the 33Fe@C/Al (Fig. S2b†) composite already few particles bigger than 10 nm are visible with particle sizes ranging from 2.2 to 19.0 nm. Moreover, although for the 25Fe@C/Al and 15Fe@C/Al composites the PSD could not be estimated, from the TEM pictures a larger size of the Fe particles is appreciated. These results show that a high fraction of boehmite increases the Fe nanoparticle particle size which goes in line with the results obtained by PXRD.
 | ||
| Fig. 5 TEM micrographs of 38Fe@C (a), 33Fe@C/Al (b), 25Fe@C/Al (c) and 15Fe@C/Al (d). | ||
3.2. HT-FTS catalytic performance
Not only the scope of this study is to create mechanically strong catalysts, but also highly active and selective towards light olefin in HT-FTS. Therefore, all the samples have been tested in HT-FTS and their performance has been evaluated in terms of catalytic activity and selectivity and catalytic and mechanical stability.In order to evaluate and compare the activity and stability of the pure 38Fe@C and the 33Fe@C/Al, 25Fe@C/Al and 15Fe@C/Al composites during 50 h TOS, the degree of dilution of the catalytic bed needed to be modified to avoid blocking of the reactors. When the same dilution of catalyst was employed in the reaction, the reactors started collapsing in the following order: 38Fe@C (at TOS = 3 h), 33Fe@C/Al (at TOS = 22 h) and then the reaction was stopped at 50 h TOS for 25Fe@C/Al and 15Fe@C/Al. This evidences that the reactors collapsed according to the boehmite loading in the corresponding catalytic bed. In this manner, the higher the amount of binder in the composite, the less dilution was required, thus demonstrating the role of boehmite in improving the mechanical resistance of the catalyst in the HT-FTS process.
When a low concentration of binder is used as in the case of 33Fe@C/Al the steady state conversion in HT-FTS is the same as that of 38Fe@C (Fig. 6a). Increasing the amount of binder, as in 25Fe@C/Al and 15Fe@C/Al, results in a considerable decreased CO conversion. Based on the characterization results the activity loss is mainly attributed to an increase in the Fe crystallite size for the composites with high binder loading as stated by XRD/TEM analysis.
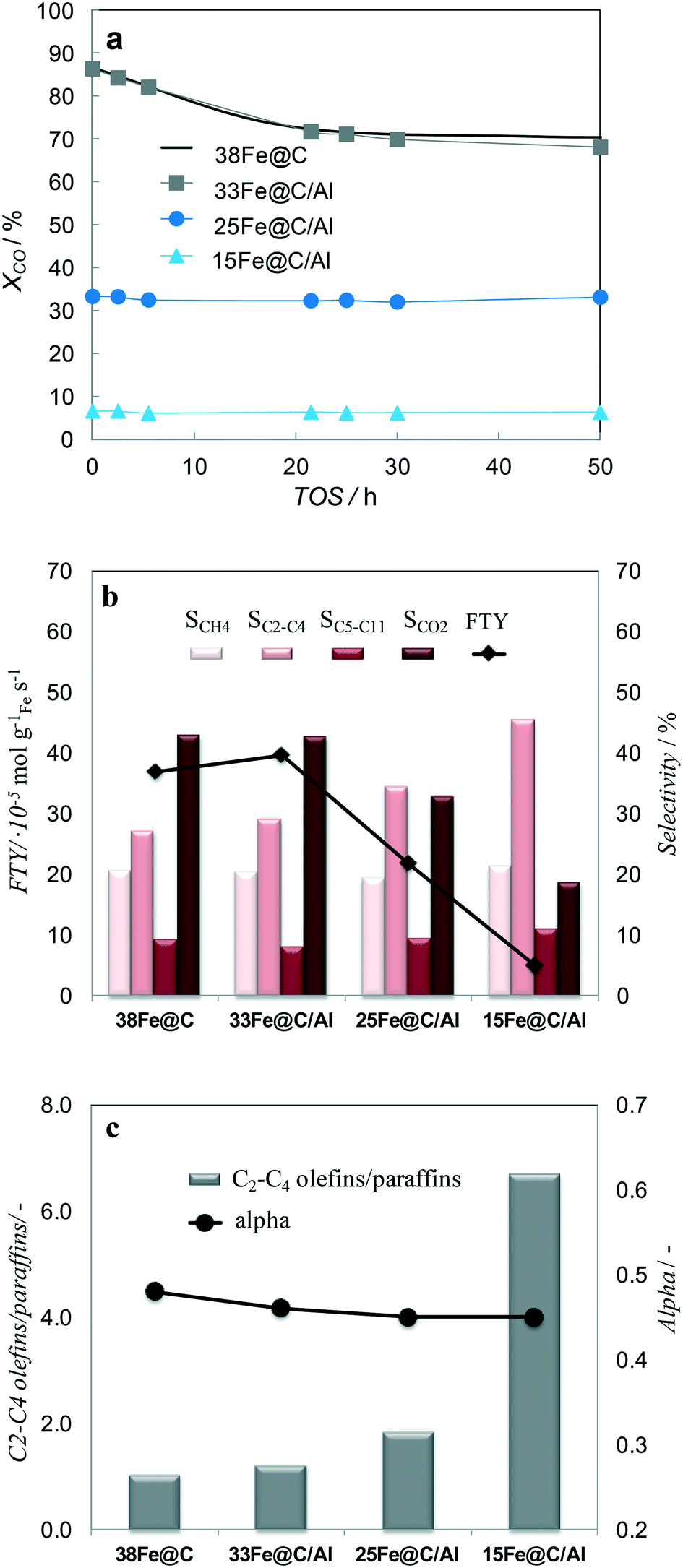 | ||
| Fig. 6 TOS evolution of XCO for all the catalysts (a) XCO and product selectivities (eqn (4)) after 50 h TOS (b) and alpha values and O/P ratio of short chain (C2–C4) hydrocarbons after 50 h TOS (c). HT-FTS conditions are 340 °C, 15 bar, H2/CO = 1, GHSV = 55 mmolCO,STP gFe−1 min−1. | ||
Regarding the stability of the catalysts, the activity for 38Fe@C and 33Fe@C/Al slightly decreases during the first 20 h TOS. From 20 h up to 50 h TOS the activity remains practically the same. In the case of the 25Fe@C/Al and 15Fe@C/Al zero time on stream activity remains the same during the 50 h TOS, thus highlighting the stability of these materials.
To analyze the selectivity to different products in HT-FTS, the obtained product distribution after 50 h TOS is shown in Fig. 6b for all the catalysts. Although to a lesser extent than for HT-FTS, the activity for the WGS reaction decreases with the amount of binder, as evidenced by the lower CO2 selectivity obtained for the composites with the lowest Fe loadings. Therefore, leaving CO2 formation and the WGS functionality aside and talking in terms of FTY to describe the catalytic activity, differences between the pure 38Fe@C and the composites become smaller than when using overall CO conversion. Nevertheless, despite the lower FTY of the 25Fe@C/Al and 15Fe@C/Al composites, their productivity is still higher than that of commercial benchmark catalysts, such as the well-known Ruhrchemie (FTY = 4.90 × 10−6 molCO gFe−1 s−1).7
The correlation of methane selectivity with CO conversion is not straightforward for Fe-based FT catalysts.38 Remarkably, despite differences in CO conversion, the CH4 selectivity is similar for all the catalysts and so is the selectivity to C5–C11 hydrocarbons. Concerning the chain length of products, alpha values are also very similar for all the catalysts (Fig. 6c). The main difference lies in the overall selectivity to C2–C4 hydrocarbons, which considers both saturated and unsaturated short chain hydrocarbons and increases for higher loadings of binder. Moreover, the selectivity to lower C2–C4 olefins for 38Fe@C (SC2–C4= = 14%), 33Fe@C/Al (SC2–C4= = 16%), 25Fe@C/Al (SC2–C4= = 22%) and 15Fe@C/Al (SC2–C4= = 39%) substantially increases when increasing the Al content in the composites (Fig. 6c). As a result, differences in the yield of C2–C4 olefins get smaller between the 38Fe@C (YC2–C4= = 9.8%) catalyst and the 33Fe@C/Al (YC2–C4= = 10.9%), 25Fe@C/Al (YC2–C4= = 7.4%), 15Fe@C/Al (YC2–C4= = 2.5%) composites.
As displayed above, differences in conversion are significant under the same HT-FTS operating conditions for the pure MOF derived catalyst and the MOF/AlOOH derived composites here studied. Therefore, in order to fairly analyze the effect of the binder on the final product distribution and towards lower olefins, the behaviour in HT-FTS has been studied at iso-conversion levels for all the catalysts and it is shown in Table 2. To this purpose, using the same operating conditions (340 °C, 15 bar and H2/CO = 1) but higher GHSV, the conversion of the 38Fe@C has been decreased to match the same levels of those obtained with the 25Fe@C/Al and the 15Fe@C/Al composites. At approximately similar conversions, there are two main effects visible when adding the inorganic binder to the catalyst: a lower selectivity towards CO2 and a higher C2–C4 selectivity with a large increase in the C2–C4 olefin/paraffin ratio (RC2–C4=/nC2–C4). For instance, at 33% CO conversion 25Fe@C/Al presents lower CO2 selectivity (SCO2 = 33%), higher selectivity to light hydrocarbons (SC2–C4= = 34%) together with a high olefin/paraffin ratio (RC2–C4=/nC2–C4 = 1.9) than the 38Fe@C/Al catalyst (SCO2 = 40%, SC2–C4 = 28% and RC2–C4=/nC2–C4 = 1.4). The lower CO2 selectivity is due to lower WGS activity when using the composites, indicating that more water and less hydrogen are available in the reaction medium, which increases the alpha or the chain length of the hydrocarbons. On the other hand, the increase in lower olefin selectivity could be due to either the presence of the coordinatively unsaturated Al sites within the nanocomposites or to the increasing Fe crystallite size in the catalysts, as discussed below.
| X CO, % | Catalyst | GHSV, mmolCO,STP gFe−1 min−1 | S C2–C4, % | S CO2, % | α | R C2–C4=/nC2–C4 |
|---|---|---|---|---|---|---|
| 70 | 38Fe@C | 55 | 27 | 43 | 0.48 | 1 |
| 68 | 33Fe@C/Al | 55 | 29 | 43 | 0.46 | 1.2 |
| 38 | 38Fe@C | 117 | 28 | 40 | 0.43 | 1.2 |
| 33 | 25Fe@C/Al | 55 | 34 | 33 | 0.49 | 1.9 |
| 9 | 38Fe@C | 411 | 34 | 29 | 0.42 | 2.3 |
| 7 | 15Fe@C/Al | 55 | 45 | 19 | 0.45 | 6.7 |
| 9 | 0.6K–38Fe@C | 694 | 28 | 39 | 0.56 | 6.9 |
Likewise, potassium is a well-known promoter in the Fe catalysed HT-FTS. It increases the adsorption of CO and consequently decreases the relative amount of adsorbed hydrogen thus hindering the hydrogenation of secondary alkenes.39 In short, K addition boosts both activity and olefin selectivity in HT-FTS. In view of the latter effect, the behaviour of the 15Fe@C/Al composite catalyst has been compared to that of a K-promoted MOF-derived 38Fe@C catalyst previously reported.7 At low CO conversion, both the K-promoted and the 15Fe@C/Al composite afford similar olefin/paraffin ratio (RC2–C4=/nC2–C4 = 6.9 and 6.7, respectively). However, CO2 selectivity for the K-promoted catalyst (SCO2 = 39%) is twice that of the 15Fe@C/Al (SCO2 = 19%) composite and at the same time, the selectivity towards C2–C4 range hydrocarbons is almost half (SC2–C4 = 28%) of that obtained with the 15Fe@C/Al composite (SC2–C4 = 45%), meaning that the overall selectivity to C2–C4 olefins is higher with the 15Fe@C/Al.
All in all, the two most remarkable effects in the MOF derived Fe@C catalysts for HT-FTS when adding AlOOH as inorganic binder are: i) an increase in the mechanical resistance, thus in the durability of the catalysts and ii) an increase in the selectivity to lower olefins.
Since the mechanical resistance of a material is directly correlated to its porosity40 and in order to study how the addition of AlOOH enhances the durability of the composites, the evolution of the textural properties of the 15Fe@C/Al composite have been measured and compared to those of the pure 38Fe@C. N2 adsorption–desorption measurements were conducted after the preliminary bed integrity test (FT-PT test) and after 3 and 50 h of HT-FTS catalytic runs for the pure 38Fe@C and the 15Fe@C/Al composite, respectively. These results are shown in Table S3.†
S BET for the pure MOF derived catalyst and the composite slightly varies after the bed integrity test performed under N2 flow and 15 bar for 3 h. Nevertheless, after running HT-FTS for 3 h, the SBET of pure 38Fe@C decreases from 274 to 57 cm2 g−1 whereas for the 15Fe@C/Al the decrease is considerably less shifting from 160 to 112 cm2 g−1 after 50 h TOS in HT-FTS. Regarding the pore volume, the mesoporosity of the 15Fe@C/Al composite remains almost the same after HT-FTS. Nevertheless, the total pore volume of 38Fe@C/Al catalyst drops down to 0.24 cm3 g−1 in the mechanical test and down to 0.073 cm3 g−1 after only 3 h TOS in HT-FTS and its mesoporosity decreases about 40% after 5 h of the FT-PT bed integrity test and 73% after 3 h TOS in HT-FTS. Additionally, the TGA profiles in Fig. S3† show that no deactivation due to carbon deposition takes place during 50 h TOS, since there is hardly variation in the carbon content for the fresh and the spent catalysts. It is also noteworthy that the TGA profiles of the composites with less binder shift to the right towards higher temperature, which is probably linked to the aforementioned collapse of the porous structure. Overall, these results reinforce the fact that the addition of AlOOH to the catalyst helps in preserving the initial textural properties of the composite, improving the durability of the catalyst in the reaction.
To gain insight into the role of the binder on the activity loss in the HT-FTS process, two additional materials have been synthesized and compared with the 25Fe@C/Al composite. These additional samples have been prepared using the starting 70/30 Fe–BTC/AlOOH mass ratio. The first material consists in the pure carbonized MOF subsequently mixed with the boehmite thus avoiding possible interactions between the Fe and Al during the pyrolysis (11.5Fe@C/Al). The second material has been carbonized at a lower temperature of 400 °C (10.5Fe@C/Al) thus minimizing sintering of the Fe nanoparticles.7 The iron loading for these two materials is similar, being 11.5 and 10.5 wt% for the non-carbonized and the carbonized at 400 °C, respectively. These two new materials have been tested in HT-FTS under the same operating conditions as in Fig. 6.
With the 10.5Fe@C/Al catalyst, the activity in HT-FTS is slightly lower than that obtained when the composite is pyrolyzed at 500 °C (Fig. S4†). As a matter of fact, when decreasing the pyrolysis temperature down to 400 °C an induction period is required to reach stable performance of the 10.5Fe@C/Al catalyst. These results go in line with previous reported work where the low activity was attributed to lower porosity of the carbon matrix due to incomplete removal of the bulky pyrolysis products from the micropores at 400 °C.21 On the other hand, for the 11.5Fe@C/Al composite prepared with the pyrolyzed MOF and the fresh boehmite, the initial activity is similar as that obtained with the pure 38Fe@C. Since the composite is not heat treated (11.5Fe@C/AlOOH), the activity drastically decreases and the reactor got plugged after 30 h TOS. Hence, the mechanical stability of the catalyst are negatively affected when AlOOH is not heat treated together with the Fe–BTC. Altogether, these results point out that the lower activity for higher amount of binder is not only linked to the Fe particle size variation in the composites but also to possible Fe–Al interactions created during the pyrolysis, which although not detected by PXRD may still be present. The Fe–Al interactions may hinder carburization of Fe towards its transformation into Hägg carbide (χ-Fe5C2) during the activation period in HT-FTS,8 leading to a lower overall activity.
At iso-conversion conditions, the light olefin selectivity increases for composites with a higher amount of binder. Although disclosing this mechanism is beyond the aim of this study, possible explanations may be the presence of coordinatively unsaturated Al sites (Alcus) and/or the increasing size of Fe crystallites with the amount of binder:
a) After pyrolysis, the presence of coordinatively unsaturated Al sites (Alcus) in the Fe@C/Al composites increases with higher amounts of binder, as evidenced by IR of pyridine adsorption. The amount of Alcus sites increases indeed with the amount of AlOOH employed to prepare the composite and so does the olefin selectivity in HT-FTS. Extra framework aluminum moieties are thought to play a significant role for dehydrogenation of light paraffins.41 Very recently Rodemerck et al.42 have proven that bare alumina shows high activity in dehydrogenation of isobutane to isobutylene. The authors relate the activity to surface coordinatively unsaturated Al sites (Alcus), which are generated upon removal of OH groups during alumina treatment at high temperatures. In such wise, Alcus and neighbouring lattice oxygen represent the active site for isobutane dehydrogenation. However, dehydrogenation of light paraffins is a thermodynamically limited and highly endothermic reaction, which, according to Le Chatelier's principle, implies that higher reaction temperature and/or lower paraffin and hydrogen partial pressures are needed to achieve high conversions. Indeed, temperatures of 550−750 °C are typically required in the dehydrogenation of C2−C4 paraffins to obtain alkane conversions ≥50% at 1 bar.43
b) Other possible explanation is that given by Gallegos et al.44 who attribute the enhanced olefin selectivity in HT-FTS over Fe/SiO2–MgO catalysts to an increased Fe crystallite size. Likewise, Xie et al.45 studied the fundamentals of structure sensitivity and promoter effects in HT-FTS by steady state isotopic transient kinetic analysis. These authors claim that H-coverage on supported iron carbide particles displays a negative correlation with lower olefin selectivity and that H-coverage decreases with increasing Fe particle size in HT-FTS, thus indicating that bigger Fe particles are more selective towards boost light olefin formation in the process.
Conclusions
After pyrolysis in N2 atmosphere at 500 °C, the Fe–BTC MOF is transformed into a mixture of Fe species embedded in a porous carbon matrix (38Fe@C). Likewise, co-pyrolysis of Fe–BTC and mesoporous AlOOH at 500 °C leads to mixed mesoporous Fe@C/γ-Al2O3 materials consisting of homogeneously dispersed carbon-embedded Fe nanoparticles on γ-Al2O3. These Fe@C/Al composites feature some intriguing properties such as i) enhanced mesoporosity as a result of mixing the microporous MOF derived Fe@C with the purely mesoporous AlOOH derived Al2O3, ii) higher Fe/Fe3O4 ratio after exposure to air, iii) bigger Fe nanoparticle size due to partial gasification of the carbon by the water released from AlOOH in the pyrolysis, iv) the presence of coordinatively unsaturated aluminium cations, Alcus3+ present in the Al2O3 obtained from AlOOH and iv) lower reducibility of iron species in the final catalyst as a consequence of water released during the reduction step which retards the reduction. The Fe@C/Al composites show good mechanical resistance in HT-FTS thus improving the catalytic and mechanical stability of Fe@C catalysts in the process. Although the activity decreases for high loading of Al2O3 in the composites, the selectivity to the targeted C2–C4 olefin fraction does increase. Possible causes for the increased light olefin selectivity are thought to be the presence of AlCUS3+ Lewis acid sites that promote dehydrogenation reaction and/or the bigger Fe crystals that exhibit a reduced H-coverage on the active Fe sites thus promoting olefin formation. Furthermore, the selectivity to C2–C4 of the composite is better than that of the K-promoted 38Fe@C and additionally produces less CO2. In view of these results and considering that the optimum catalyst should meet a compromise between high activity, high selectivity to lower olefins and high chemical and mechanical stability, the Fe–BTC/AlOOH ratio ranging from 70/30 to 90/10 is proposed to lead to the best Fe@C/Al composite for HT-FTS. To sum up, we synthesized highly resistant Fe@C–Al composites that have a more competitive product slate for HT-FTS operation than state-of-the-art catalysts driving to obtain lower olefins.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 704473.References
- I. Amghizar, L. A. Vandewalle, K. M. Van Geem and G. B. Marin, Engineering, 2017, 3, 171–178 CrossRef.
- H. M. Torres Galvis and K. P. de Jong, ACS Catal., 2013, 3, 2130–2149 CrossRef CAS.
- A. Corma, F. V. Melo, L. Sauvanaud and F. Ortega, Catal. Today, 2005, 107–108, 699–706 CrossRef CAS.
- L. Zhong, F. Yu, Y. An, Y. Zhao, Y. Sun, Z. Li, T. Lin, Y. Lin, X. Qi, Y. Dai, L. Gu, J. Hu, S. Jin, Q. Shen and H. Wang, Nature, 2016, 538, 84–87 CrossRef CAS PubMed.
- H. M. Torres Galvis, A. C. J. Koeken, J. H. Bitter, T. Davidian, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Catal. Today, 2013, 215, 95–102 CrossRef CAS.
- K. Cheng, M. Virginie, V. V. Ordomsky, C. Cordier, P. A. Chernavskii, M. I. Ivantsov, S. Paul, Y. Wang and A. Y. Khodakov, J. Catal., 2015, 328, 139–150 CrossRef CAS.
- V. P. Santos, T. A. Wezendonk, J. J. D. Jaén, A. I. Dugulan, M. A. Nasalevich, H.-U. Islam, A. Chojecki, S. Sartipi, X. Sun, A. A. Hakeem, A. C. J. Koeken, M. Ruitenbeek, T. Davidian, G. R. Meima, G. Sankar, F. Kapteijn, M. Makkee and J. Gascon, Nat. Commun., 2015, 6, 6451 CrossRef CAS PubMed.
- T. A. Wezendonk, Q. S. E. Warringa, V. P. Santos, A. Chojecki, M. Ruitenbeek, G. Meima, M. Makkee, F. Kapteijn and J. Gascon, Faraday Discuss., 2017, 197, 225–242 RSC.
- J. Sun, Y. Chen and J. Chen, Catal. Commun., 2017, 91, 34–37 CrossRef CAS.
- L. Oar-Arteta, A. Remiro, A. T. Aguayo, M. Olazar, J. Bilbao and A. G. Gayubo, J. Ind. Eng. Chem., 2016, 36, 169–179 CrossRef CAS.
- P. Pérez-Uriarte, M. Gamero, A. Ateka, M. Díaz, A. T. Aguayo and J. Bilbao, Ind. Eng. Chem. Res., 2016, 55, 1513–1521 CrossRef.
- M. Ray, P. Bhattacharya, R. Das, K. Sondhi, S. Ghosh and S. Sarkar, J. Porous Mater., 2015, 22, 1043–1052 CrossRef CAS.
- X. Bokhimi, J. A. Toledo-Antonio, M. L. Guzmán-Castillo, B. Mar-Mar, F. Hernández-Beltrán and J. Navarrete, J. Solid State Chem., 2001, 161, 319–326 CrossRef CAS.
- S. Jang, S. W. Kang, D. H. Chun, H.-T. Lee, J.-I. Yang, H. Jung, H.-D. Jeong, K. M. Nam and J. C. Park, New J. Chem., 2017, 41, 2756–2763 RSC.
- H. M. Torres Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835–838 CrossRef CAS PubMed.
- P. Scherrer, Math.-Phys. Kl., 1918, 2, 98–100 Search PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- M. M. Dubinin, E. D. Zaverina and L. V. Radushkevich, Zh. Fiz. Khim., 1947, 21, 1351–1362 CAS.
- K. Kaneko and C. Ishii, Colloids Surf., 1992, 67, 203–212 CrossRef CAS.
- S. Sartipi, H. Jansma, D. Bosma, B. Boshuizen, M. Makkee, J. Gascon and F. Kapteijn, Rev. Sci. Instrum., 2013, 84, 124101 CrossRef PubMed.
- T. A. Wezendonk, V. P. Santos, M. A. Nasalevich, Q. S. E. Warringa, A. I. Dugulan, A. Chojecki, A. C. J. Koeken, M. Ruitenbeek, G. Meima, H.-U. Islam, G. Sankar, M. Makkee, F. Kapteijn and J. Gascon, ACS Catal., 2016, 6, 3236–3247 CrossRef CAS.
- A. R. Oveisi, A. Khorramabadi-zad and S. Daliran, RSC Adv., 2016, 6, 1136–1142 RSC.
- Y. Liu, D. Ma, X. Han, X. Bao, W. Frandsen, D. Wang and D. Su, Mater. Lett., 2008, 62, 1297–1301 CrossRef CAS.
- H. Zhang, P. Li, W. Cui, C. Liu, S. Wang, S. Zheng and Y. Zhang, RSC Adv., 2016, 6, 27235–27241 RSC.
- T. A. Wezendonk, V. P. Santos, M. A. Nasalevich, Q. S. E. Warringa, A. I. Dugulan, A. Chojecki, A. C. J. Koeken, M. Ruitenbeek, G. Meima, H.-U. Islam, G. Sankar, M. Makkee, F. Kapteijn and J. Gascon, ACS Catal., 2016, 3236–3247, DOI:10.1021/acscatal.6b00426.
- W. Xia, V. Hagen, S. Kundu, Y. Wang, C. Somsen, G. Eggeler, G. Sun, G. Grundmeier, M. Stratmann and M. Muhler, Adv. Mater., 2007, 19, 3648–3652 CrossRef CAS.
- C.-W. Lee, H.-S. Kang, Y.-H. Chang and Y.-M. Hahm, Korean J. Chem. Eng., 2000, 17, 266–272 CrossRef CAS.
- T. Armaroli, D. Minoux, S. Gautier and P. Euzen, Appl. Catal., A, 2003, 251, 241–253 CrossRef CAS.
- C. Morterra, G. Magnacca, G. Cerrato, N. Del Favero, F. Filippi and C. V. Folonari, J. Chem. Soc., Faraday Trans., 1993, 89, 135–150 RSC.
- P. Raybaud, M. Digne, R. Iftimie, W. Wellens, P. Euzen and H. Toulhoat, J. Catal., 2001, 201, 236–246 CrossRef CAS.
- H. Leclerc, A. Vimont, J.-C. Lavalley, M. Daturi, A. D. Wiersum, P. L. Llwellyn, P. Horcajada, G. Ferey and C. Serre, Phys. Chem. Chem. Phys., 2011, 13, 11748–11756 RSC.
- S. Wuttke, P. Bazin, A. Vimont, C. Serre, Y.-K. Seo, Y. K. Hwang, J.-S. Chang, G. Férey and M. Daturi, Chem. – Eur. J., 2012, 18, 11959–11967 CrossRef CAS PubMed.
- A. Vimont, H. Leclerc, F. Maugé, M. Daturi, J.-C. Lavalley, S. Surblé, C. Serre and G. Férey, J. Phys. Chem. C, 2007, 111, 383–388 CAS.
- Y. Wu, H. Yu, F. Peng and H. Wang, Mater. Lett., 2012, 67, 245–247 CrossRef CAS.
- O. J. Wimmers, P. Arnoldy and J. A. Moulijn, J. Phys. Chem., 1986, 90, 1331–1337 CrossRef CAS.
- Z. Hu, W.-Z. Li, K.-Q. Sun and B.-Q. Xu, Catal. Sci. Technol., 2013, 3, 2062–2071 CAS.
- X. Zhang, Y. Ge, S.-P. Hannula, E. Levanen and T. Mantyla, J. Mater. Chem., 2009, 19, 1915–1922 RSC.
- J. Yang, W. Ma, D. Chen, A. Holmen and B. H. Davis, Appl. Catal., A, 2014, 470, 250–260 CrossRef CAS.
- L. A. Cano, A. A. Garcia Blanco, G. Lener, S. G. Marchetti and K. Sapag, Catal. Today, 2017, 282(Part 2), 204–213 CrossRef CAS.
- T.-J. Ha, H.-H. Park, E. S. Kang, S. Shin and H. H. Cho, J. Colloid Interface Sci., 2010, 345, 120–124 CrossRef CAS PubMed.
- T. F. Narbeshuber, A. Brait, K. Seshan and J. A. Lercher, J. Catal., 1997, 172, 127–136 CrossRef CAS.
- U. Rodemerck, E. V. Kondratenko, T. Otroshchenko and D. Linke, Chem. Commun., 2016, 52, 12222–12225 RSC.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- N. G. Gallegos, A. M. Alvarez, M. V. Cagnoli, J. F. Bengoa, S. G. Marchetti, R. C. Mercader and A. A. Yeramian, J. Catal., 1996, 161, 132–142 CrossRef CAS.
- J. Xie, J. Yang, A. I. Dugulan, A. Holmen, D. Chen, K. P. de Jong and M. J. Louwerse, ACS Catal., 2016, 6, 3147–3157 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cy01753g |
| This journal is © The Royal Society of Chemistry 2018 |