
Attractive non-DLVO forces induced by adsorption of monovalent organic ions
Alexander M.
Smith
 ,
Plinio
Maroni
and
Michal
Borkovec
*
,
Plinio
Maroni
and
Michal
Borkovec
*
Department of Inorganic and Analytical Chemistry, University of Geneva, Sciences II, 30 Quai Ernest-Ansermet, 1205 Geneva, Switzerland. E-mail: michal.borkovec@unige.ch
First published on 30th November 2017
Abstract
Direct force measurements between negatively charged colloidal particles were carried out using an atomic force microscope (AFM) in aqueous solutions containing monovalent organic cations, namely tetraphenylarsonium (Ph4As+), 1-hexyl-3-methylimidazolium (HMIM+), and 1-octyl-3-methylimidazolium (OMIM+). These ions adsorb to the particle surface, and induce a charge reversal. The forces become attractive at the charge neutralization point, but they are stronger than van der Waals forces. This additional and unexpected attraction decays exponentially with a decay length of a few nanometers, and is strikingly similar to the one previously observed in the presence of multivalent ions. This attractive force probably originates from coupled spontaneous charge fluctuations on the respective surfaces as initially suggested by Kirkwood and Shumaker.
Introduction
Classical theory of Derjaguin, Landau, Verwey, and Overbeek (DLVO) has proven to be surprisingly successful to quantify forces acting between liquid–solid interfaces in various electrolyte solutions.1–5 This evidence is based on a continuously increasing body of direct force measurements using an atomic force microscope (AFM), a surface force apparatus, optical tweezers, or a total internal reflection microscope.4–16 These techniques reveal that the measured force profiles can be modeled extremely well by superposing van der Waals and double layer forces, as surmised by the DLVO theory. In some situations, however, the double layer forces must be described by the full Poisson–Boltzmann (PB) theory and charge regulation effects must be considered, especially when dealing with dissimilar surfaces.3,12–14,17 Thereby, the diffuse layer potential and regulation properties of the interface must be extracted from the experimental force profiles. Predicting these quantities from more detailed interfacial models seems challenging and remained unreliable so far. However, when these parameters are extracted from the force profiles, these profiles can be described in an accurate and robust fashion down to nm-distances, even in salt solutions containing multivalent ions.6,18,19The applicability of DLVO theory in the presence of multivalent ions may seem surprising since several researchers have reported that the underlying PB theory breaks down near charged interfaces in such systems due to the neglect of ion–ion correlations.20–22 However, this assertion only applies to small distances from the interface and assuming that the (bare) surface potential remains unaltered.6,23 Multivalent ions typically adsorb to oppositely charged interfaces, whereby they modify the surface charge density, and therefore they influence the diffuse layer potential strongly. When such (effective) diffuse layer potentials are used in PB theory, the ionic and force profiles can be accurately described down to several nanometers.
Inspection of the currently available force data in various multivalent electrolyte solutions reveals one characteristic deviation from DLVO theory, however. Multivalent counterions of valence higher than 2 systematically induce a charge reversal and near the charge reversal point forces acting between two similar interfaces become attractive.6,18,24 This feature can be qualitatively understood from DLVO theory, since at the charge reversal point the double layer force vanishes and the force profile is dominated by the attractive van der Waals force. However, the experimentally measured attractive forces are substantially stronger than van der Waals forces, and they disagree with DLVO theory.18,19,25 The additional non-DLVO contribution can be fitted with an exponential profile with a decay length of a few nanometers. A possible interpretation of this additional force could be due to ion–ion correlation effects.20 Consideration of these effects leads to an additional attractive force, and so far, this additional attraction has been exclusively observed in the presence of multivalent ions.
The purpose of this article is to show that this additional attractive non-DLVO force is unrelated to ionic valences, and that very similar attraction can be observed in the presence of monovalent organic ions. The respective direct force measurements are carried out with the colloidal probe technique based on the AFM. Forces between two types of negatively charged colloidal particles and three types of organic monovalent cations are investigated. Attractive non-DLVO forces are observed in all systems studied, and these forces strongly resemble those observed in systems containing multivalent ions.
Experimental techniques and methods
Materials
Polystyrene latex particles functionalized with sulfate groups (Invitrogen Corp.) with an average diameter of 2.9 μm were dialyzed with a cellulose ester membrane against ultrapure water until the conductivity value was less than 70 μS m−1.18 The silica particles with a diameter of 5.2 μm were purchased from Bangs Laboratories Inc., USA. Electrolyte solutions of potassium chloride (AcrosOrganics, 99+%), tetraphenylarsonium chloride (Ph4AsCl, Sigma-Aldrich, 97%), 1-hexyl-3-methylimidazolium chloride (HMIMCl, C6C1ImCl) and 1-octyl-3-methylimidazolium chloride (OMIMCl, C8C1ImCl, Iolitec, >98%) solutions were prepared using ultrapure water and adjusted to pH 4.0 with hydrochloric acid.Direct force measurements
Forces between pairs of similar colloidal particles were measured using a closed-loop AFM (MFP-3D, Asylum Research) mounted on an inverted optical microscope (Olympus IX 73). Just prior to introduction into the AFM fluid cell, the solutions were degassed in vacuum for 10 min under stirring and filtered with a 0.1 μm syringe filter. Especially at lower concentrations the experimental results were the same as without degassing, and under these conditions the degassing step was skipped. After centering a pair of particles using an optical microscope, about 100 approach-retraction cycles were measured at a velocity of about 300 nm s−1. The deflection signal was converted to force profiles by subtracting the baseline and the constant compliance region, and by considering the respective spring constants of 0.2–0.5 N m−1. This constant was measured using the method developed by Sader et al.,26 which relies on lateral dimensions of the cantilever and its frequency response. Forces obtained from repeated approach curves were averaged, leading to a force resolution of about 2 pN down to distances of about 0.3 nm.Prior to the force measurements, the glass plate of the fluid cell was cleaned in piranha solution then functionalized with 3-(ethoxydimethylsilyl)propylamine via vapor deposition. The substrate was mounted in the fluid cell, rinsed with pure water, and a particle suspension was introduced. Thereby, the sulfate latex particles attach to the positively charged substrate. A similarly functionalized tip-less cantilever (MicroMash) was used to press and pick up particles under liquid to avoid exposing the hydrophobic particles to air.
The silica particles (Bangs Laboratories Inc., USA) with a diameter of 5.2 μm were attached to tipless cantilevers with epoxy glue in air. Particles were also sprinkled over quartz substrates and then heat-treated together with the cantilevers at 1150 °C for 3 hours. This sintering process serves not only to firmly attach the particles, but also shrinks the particles to 4.4 μm, reduces the surface roughness, and removes any traces of glue.27 Immediately prior to force measurements, silica substrates and cantilevers were rinsed thoroughly with ethanol, subsequently with ultrapure water, then dried, and cleaned in air plasma for 20 minutes.
Previous studies found that the root mean square roughness of similarly prepared latex and silica particles was 0.8 nm and 0.7 nm, respectively.18,27
Modeling of interaction forces
Interaction forces between particles are calculated within an extended DLVO theory, and modeled as the sum| FDLVO = FvdW + Fdl + Fatt | (1) |
The dependence of van der Waals force on the separation distance h is modeled with the non-retarded theory and invoking the Derjaguin approximation1
 | (2) |
The double layer force is evaluated from the swelling pressure acting between two charged plates, for which the PB equation is solved numerically.28 In a monovalent electrolyte, the electric potential ψ(x) satisfies the equation
 | (3) |
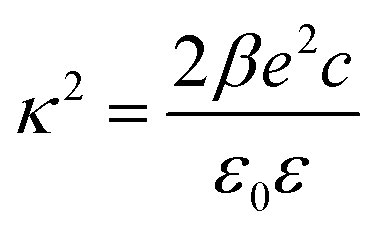 | (4) |
 | (5) |
 | (6) |
 | (7) |
 | (8) |
The double layer force is obtained by calculating the disjoining pressure originating from the double layer from the relation
 | (9) |
 | (10) |
An additional attractive non-DLVO contribution is considered in the calculations, and this force is modeled with an exponential profile, namely18,19
 | (11) |
Results and discussion
This article describes direct force measurements with the colloidal probe technique based on the AFM in the sphere–sphere geometry (Fig. 1). We investigate two types of negatively charged colloidal particles and three types of organic cations as shown in Fig. 1. These cations include tetraphenylarsonium (Ph4As+), 1-hexyl-3-methylimidazolium chloride (HMIM+), and 1-octyl-3-methylimidazolium chloride (OMIM+). All these cations strongly adsorb to both types of particles, and thereby they induce a charge reversal. Additional attractive non-DLVO forces are observed near the charge reversal point in all systems investigated, and these forces closely resemble the ones previously reported in the presence of multivalent ions. The present observations with monovalent ions thus demonstrate that such attractive non-DLVO forces are unrelated to ion–ion correlations. | ||
| Fig. 1 Optical micrograph and scheme showing the experimental sphere–sphere interaction geometry of particles attached to the substrate and cantilever (left). Structural formulas of the cations investigated (right). | ||
Generic features
The organic ions studied adsorb strongly to water–solid interfaces due to their hydrophobic character. Since these ions and the substrate are oppositely charged, they induce a charge reversal of the substrate. This charge reversal can be evidenced by direct force measurements in solutions of the chloride salt of the tetraphenylarsonium cation (Ph4As+) and negatively charged sulfate latex particles of 2.9 μm in diameter (Fig. 2). We report the forces F normalized by the effective radius Reff, thus F/Reff. At low concentrations of the Ph4As+ cation, forces are strongly repulsive and long-ranged, indicating the importance of repulsive double-layer interactions (Fig. 2a). When the concentration of the Ph4As+ cation is increased, the magnitude of the forces decreases, while their range remains approximately the same. This behavior indicates a reduction of the magnitude of the diffuse layer potential, and signals the adsorption of the oppositely charged cations to the negatively charged particle surface. At a certain concentration, the surface becomes precisely neutralized. In the present system, the charge neutralization point occurs at a concentration of 0.25 mM of the Ph4As+ cation, and a purely attractive force is observed. When the concentration is increased further, the surface concentration of the cations increases and the surface now becomes positively charged. This aspect can be inferred from the force profile by the occurrence of the reentrant repulsive double layer force, whose magnitude now increases with increasing concentration (Fig. 2b). As the concentration is increased even further, the range of the repulsive force decreases due to screening (Fig. 2c). At high concentrations, the force becomes attractive again. This attractive force is dominated by van der Waals forces. Comparison of the latter attractive force profile with the profile with the one observed at the neutralization point reveals that the latter force is more attractive, suggesting the presence of an additional attractive non-DLVO force near the charge neutralization point. | ||
| Fig. 2 Measured force profiles between sulfate latex particles across aqueous Ph4AsCl solutions at pH 4.0 compared with DLVO theory within the constant regulation approximation (dashed line) and including additional non-DLVO exponential attraction (full line). The regulation parameter and the decay length of the attraction are fixed and given in Table 1. The force profiles are shown for concentrations of Ph4AsCl (b) ≤0.25 mM, (c) ≥0.25 mM and ≤1.0 mM, and (d) ≥1.0 mM. | ||
Quantitative interpretation
To gain better insights into the underlying mechanisms governing these forces, the force profiles were quantitatively interpreted with extended DLVO theory. The DLVO theory considers the classical non-retarded van der Waals force and the double layer force, which are calculated by means of the full PB equation. The force profile obtained from this theory is further modified by means of an additive, attractive exponential force.Best fits with this model are shown in Fig. 2, whereby the results of the DLVO theory are compared with the ones in the presence of the attractive non-DLVO force. The entering parameters were determined as follows. From the force profile at a high concentration, we extract H = (3.3 ± 0.3) × 10−21 J. This value of the Hamaker constant compares well with the one measured for similar latex particles earlier.6,18 One should note that the present value is somewhat smaller than the one obtained from spectroscopic data theoretically.15 These deviations are probably caused by minor surface roughness. Once the Hamaker constant is found, the remaining parameters of the model, namely the diffuse layer potential ψD, the regulation parameter p, the amplitude A, and the decay length q−1 of the non-DLVO force, were determined by the least-squares fit. The electrolyte concentration was fixed to its nominal value. When this concentration was fitted, the resulting value did not deviate more than 10% from the nominal one. The regulation parameter p and the decay length q−1 of the non-DLVO force did scatter somewhat and did not show any clear trends with the concentration. These values were therefore fixed to the respective averages, namely p = 0.06 ± 0.09 and q−1 = 3.0 ± 0.2 nm (Table 1). The data were refitted by keeping these parameters constant and the resulting values of the diffuse layer potential ψD and of the amplitude A of the non-DLVO force are summarized in Fig. 3. The plot of the diffuse layer potential versus the concentration of Ph4As+ passes through zero, and then goes through a pronounced maximum (Fig. 3a). The amplitude of the non-DLVO force shows a maximum near the charge neutralization point (Fig. 3b). One should note that the sign of the diffuse layer potential cannot be directly inferred from the force profiles, as the double layer force between similarly charged surfaces is always repulsive. However, by measuring the force between the colloidal particle on the cantilever and the positively charged substrate, we can directly verify that a charge reversal has indeed occurred.
| Particles | Salt | Regulation parameter, p | Decay length, q−1 (nm) |
|---|---|---|---|
| Sulfate latex | Ph4AsCl | 0.06 ± 0.09 | 3.0 ± 0.2 |
| Sulfate latex | HMIMCl | 0.32 ± 0.10 | 1.2 ± 0.1 |
| Sulfate latex | OMIMCl | 0.40 ± 0.07 | 1.8 ± 0.2 |
| Silica | Ph4AsCl | 0.05 ± 0.05 | 3.2 ± 0.3 |
| Silica | HMIMCl | 0.45 ± 0.06 | 1.4 ± 0.3 |
| Silica | OMIMCl | 0.36 ± 0.04 | 1.9 ± 0.2 |
 | ||
| Fig. 3 Concentration dependence of different parameters obtained from best fits of the force profiles in aqueous solutions of different organic cations at pH 4.0 for sulfate latex (left column) and silica particles (right column). The solid lines serve to guide the eye only. (a) Diffuse layer potential and (b) amplitudes of non-DLVO attractive forces. Table 1 shows the corresponding regulation parameters and decay lengths, which are taken to be concentration independent. The arrows indicate the position of the charge neutralization point. | ||
We have further investigated forces in the presence of HMIM+ and OMIM+ cations. These ions induce a similar charge reversal, whereby the charge neutralization point shifts to lower concentrations with increasing hydrophobicity of the cation. For these cations, we also observed similar additional non-DLVO attractive forces. The force profiles could be quantified with the same model discussed above, albeit with different parameters. The resulting regulation parameters and decay lengths could be again fixed (Table 1). The concentration dependence of the diffuse layer potentials and the amplitude of the non-DLVO force are shown in Fig. 3. Clearly these parameters feature similar trends to the ones observed for the Ph4As+ cation. The presence of the charge reversal further agrees with recent electrophoretic mobility measurements with similar organic cations.30,31
To confirm that these features are independent of the type of particles used, we have carried out similar measurements with sintered silica particles of 4.4 μm in diameter. We extract H = (1.7 ± 0.2) × 10−21 J from the force profiles. This value matches the one reported earlier for silica particles prepared according to the same procedure.27 This value further agrees well with the theoretical estimate given by Ackler et al.32 The remaining parameters were determined in the same way as for the sulfate latex particles described above. The resulting values are summarized in Table 1 and Fig. 3. These three cations also induce a similar charge reversal for the silica particles.
We note with interest the differing adsorption order of the ions for the different particles, which are demonstrated from the respective concentration dependencies of the diffuse layer potential (Fig. 3a). For the sulfate latex particles the Ph4As+ cation adsorbs most strongly, whereas for the silica particles its adsorption strength is intermediate between HMIM+ and OMIM+. We suspect that these features are related to differing adsorption mechanisms in both cases. In the case of the hydrophobic sulfate latex particles, adsorption is probably governed by hydrophobic interactions for all three ions, and one expects the imidazolium alkyl chains to adsorb in a flat configuration. For the more hydrophilic silica particles, however, the amphiphilic HMIM+ and OMIM+ cations are expected to adsorb such that the alkyl chain will orient perpendicularly to the surface. In fact, the highest imidazolium salt concentrations shown here are close to the critical micellar concentration (CMC).33 Moreover, a short-ranged hydrophobic force is observed between silica particles acting at distances below a few nm, which indicates the presence of hydrophobic adsorbed monolayers with exposed alkyl chains.
One further observes systematic trends of the regulation parameters (Table 1). In the presence of the Ph4As+ cation, the regulation parameters are small, suggesting that the surfaces regulate close to CP conditions. The regulation parameters assume intermediate values for the HMIM+ and OMIM+ cations, which indicates that the surfaces regulate their charge less easily. Interestingly, these trends are similar for both interfaces, namely for the sulfate latex and silica. Given the fact that the variation of the diffuse layer potential with the concentration is similar for these different cations, these trends cannot be easily rationalized. The intermediate values of the regulation parameters are close to the values observed for simple salts.18,34 This resemblance suggests that co-adsorption of chloride ions could be responsible for the charge regulation of these surfaces. This aspect could also represent the reason, why the surfaces regulate more easily in the presence of the strongly adsorbing Ph4As+ cation. In this system, charge regulation could be again influenced by co-adsorption phenomena of simple ions.
Attractive non-DLVO force induced by organic monovalent ions
The presence of additional non-DLVO attractions is best demonstrated by comparing the forces observed at the charge neutralization point with the ones at high salt concentrations. The advantage of this comparison is that double layer forces are negligible under both conditions.This comparison is shown in Fig. 4. At high salt concentrations, the attractive forces are dominated by van der Waals interactions in all systems (Fig. 4a). These forces can be well described by the non-retarded van der Waals expression given in eqn (2). At the charge neutralization point, however, the forces are substantially stronger than van der Waals interactions (Fig. 4b). While their distance dependence can be well described by the additional exponential non-DLVO force given by eqn (11), these profiles are incompatible with van der Waals forces with an adjusted Hamaker constant. The respective decay lengths are summarized in Table 1. While these lengths are independent of the substrate, they depend on the type of cation. The presence of the additional attractive forces is also evident from Fig. 3b. Its amplitude goes through a maximum near the charge reversal point, while it becomes vanishingly small at high salt concentrations.
 | ||
| Fig. 4 Measured attractive force profiles compared with the van der Waals force (dashed line) and with the additional exponential non-DLVO force (full line) in aqueous solutions containing different organic cations at pH 4.0 for sulfate latex (left column) and silica particles (right column). (a) High salt concentration and (b) the charge neutralization point. The decay lengths of the additional non-DLVO exponential attraction are given in Table 1. | ||
These profiles were compared with forces measured in simple KCl solutions. At high KCl concentrations, forces in this system are attractive, and reflect the van der Waals force. At lower salt concentrations, the double layer forces induce repulsion. Since the surface of the latex particles is hydrophobic, they also feature a short-ranged attractive force of the hydrophobic origin. While this force was also surmised to be exponential, the decay length is only around 0.3 nm, which is much smaller than for the non-DLVO attractive forces described here. As already discussed earlier, this behavior is typical for monovalent salt solutions and probably is caused by short-ranged hydrophobic forces.18
Attractive non-DLVO force induced by multivalent ions
Let us now stress the analogy between the attractive non-DLVO forces in solutions of monovalent organic cations described here and the ones reported previously in the presence of multivalent ions. This comparison is again best carried out at the charge neutralization point. Fig. 5 shows a selection of previous direct force measurements between sulfate latex particles in the presence of multivalent aliphatic polyamines18 and between silica particles and a silica substrate in the presence of simpler multivalent inorganic ions.19,25 The Hamaker constants used in the calculations are 3.5 × 10−21 J for the sulfate latex and 6.0 × 10−21 J for silica.18,25 | ||
| Fig. 5 Attractive force profiles compared with the van der Waals force (dashed line) and with the additional exponential non-DLVO force (full line) in aqueous solutions containing different multivalent counterions for sulfate latex (left) and silica particles (right). The sulfate latex data were recorded in the presence of aliphatic amines with four (N4) and six (N6) amine groups at pH 4.0.18 The silica data were measured between a silica particle and a flat silica substrate in the presence LaCl3 and [Co(NH3)6]Cl3 at pH 5.5.25 The respective data in simple electrolytes are shown for comparison. The decay length of the additional non-DLVO exponential attraction is 1.0 nm for both substrates. | ||
For the multivalent ions one indeed observes very similar patterns to the ones reported here for the monovalent organic cations. At high salt concentrations, the forces are attractive and governed by van der Waals interactions. At the charge neutralization point, the forces are more strongly attractive, and the profile can be again modeled with an additional exponential term. The decay length was taken as 1.0 nm for both substrates. This number is smaller than the ones reported here, and this difference again illustrates the influence of the type of cations. However, the additional non-DLVO attractions seem less pronounced for the multivalent ions than the ones reported here, especially for the Ph4As+ cation.
Origin of the attractive non-DLVO force
Based on the similarity between additional attractive non-DLVO forces induced by multivalent ions and the organic monovalent ions investigated here, we suspect that in all situations the mechanism inducing the additional force is similar, and therefore that it must be unrelated to ionic valences. For this reason, ion–ion correlations represent an unlikely explanation of this additional force, as these correlations become increasingly important with increasing ionic valence.20–22In our view, the most likely mechanism for these additional attractions is the charge fluctuation force initially proposed by Kirkwood and Shumaker.35,36 This attractive force originates from the coupling between thermal charge fluctuations on the two interfaces. While a functional form has been proposed for two smaller macroions, there is currently little information concerning the distance dependence of this force for planar surfaces in electrolyte solutions. For this reason, we are currently unable to carry out a quantitative data analysis with this model.
Another explanation for this attraction could be due to double layer forces induced by surface charge heterogeneities.37–39 This force is expected to be exponential, and at low salt concentrations the respective decay length should be governed by the lateral size of the surface charge heterogeneities. This aspect could explain why the observed decay lengths are substantially smaller than the Debye lengths. In light of this interpretation, however, the similarities of the decay lengths for the different substrates seem surprising. Similarly, effects of surface roughness are also expected to be minor, since the observed decay lengths all exceed the root mean square values of the surface roughness. Therefore, we suspect that the origin of the observed additional forces is unrelated to surface heterogeneities and roughness effects.
A further possible interpretation of these additional forces could be in terms of hydrophobic interactions. While recent measurements of these interactions suggest that their dependence is also exponential, the respective decay lengths are situated around 0.3 nm.40,41 These small values are related to the fact that such forces originate from the structuring of the solvent near the interface.42 Such decay lengths are substantially smaller than the decay length observed here. Moreover, the presently observed additional force occurs at low concentrations of the organic cations, and disappears at higher ones. One would expect the opposite trends if this force would be of the hydrophobic origin, since the adsorbed amount of the organic cations increases with increasing concentration. For these reasons, we think that an interpretation of the additional attractive force in terms of hydrophobic interactions seems unlikely.
Conclusion
We have carried out direct force measurements between negatively charged colloidal particles in the presence of monovalent organic cations. These cations lead to charge neutralization and charge reversal. At the charge neutralization point, the forces are attractive, but they are stronger than the van der Waals force. These additional non-DLVO forces are very similar to the ones observed in the presence of multivalent ions earlier. We thus suspect that the presence of these forces is unrelated to the ionic valence, and our results point towards an origin unrelated to ion–ion correlations. These additional attractions probably result from the coupling between spontaneous charge fluctuations on the respective surfaces.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by the Swiss National Science Foundation by the projects No. 150631 and 159874 and the University of Geneva.References
- W. B. Russel, D. A. Saville and W. R. Schowalter, Colloidal Dispersions, Cambridge University Press, Cambridge, 1989 Search PubMed.
- J. Israelachvili, Intermolecular and Surface Forces, Academic Press, London, 3rd edn, 2011 Search PubMed.
- G. Trefalt, S. H. Behrens and M. Borkovec, Langmuir, 2016, 32, 380–400 CrossRef CAS PubMed.
- M. M. Elmahdy, A. Drechsler, C. Gutsche, A. Synytska, P. Uhlmann, F. Kremer and M. Stamm, Langmuir, 2009, 25, 12894–12898 CrossRef CAS PubMed.
- J. Morag, M. Dishon and U. Sivan, Langmuir, 2013, 29, 6317–6322 CrossRef CAS PubMed.
- F. J. Montes Ruiz-Cabello, G. Trefalt, P. Maroni and M. Borkovec, Langmuir, 2014, 30, 4551–4555 CrossRef CAS PubMed.
- R. M. Pashley, J. Colloid Interface Sci., 1981, 83, 531–546 CrossRef CAS.
- R. M. Pashley, J. Colloid Interface Sci., 1984, 102, 23–35 CrossRef CAS.
- V. Kuznetsov and G. Papastavrou, J. Phys. Chem. C, 2014, 118, 2673–2685 CAS.
- C. Gutsche, U. F. Keyser, K. Kegler and F. Kremer, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 76, 031403 CrossRef CAS PubMed.
- M. Nayeri, Z. Abbas and J. Bergenholtz, Colloids Surf., A, 2013, 429, 74–81 CrossRef CAS.
- R. Tivony, D. Ben Yaakov, G. Silbert and J. Klein, Langmuir, 2015, 31, 12845–12849 CrossRef CAS PubMed.
- I. Popa, P. Sinha, M. Finessi, P. Maroni, G. Papastavrou and M. Borkovec, Phys. Rev. Lett., 2010, 104, 228301 CrossRef PubMed.
- C. Zhao, D. Ebeling, I. Siretanu, D. van den Ende and F. Mugele, Nanoscale, 2015, 7, 16298–16311 RSC.
- M. A. Bevan and D. C. Prieve, Langmuir, 1999, 15, 7925–7936 CrossRef CAS.
- R. M. Espinosa-Marzal, T. Drobek, T. Balmer and M. P. Heuberger, Phys. Chem. Chem. Phys., 2012, 14, 6085–6093 RSC.
- C. Liu, E. Thormann, E. Tyrode and P. M. Claesson, J. Colloid Interface Sci., 2015, 443, 162–169 CrossRef CAS PubMed.
- M. Moazzami-Gudarzi, G. Trefalt, I. Szilagyi, P. Maroni and M. Borkovec, J. Phys. Chem. C, 2015, 119, 15482–15490 CAS.
- O. Zohar, I. Leizerson and U. Sivan, Phys. Rev. Lett., 2006, 96, 177802 CrossRef PubMed.
- L. Guldbrand, B. Jonsson, H. Wennerstrom and P. Linse, J. Chem. Phys., 1984, 80, 2221–2228 CrossRef CAS.
- R. Kjellander and S. Marcelja, Chem. Phys. Lett., 1984, 112, 49–53 CrossRef CAS.
- A. Naji, M. Kanduc, J. Forsman and R. Podgornik, J. Chem. Phys., 2013, 139, 150901 CrossRef PubMed.
- M. M. Hatlo and L. Lue, Soft Matter, 2009, 5, 125–133 RSC.
- K. Besteman, M. A. G. Zevenbergen, H. A. Heering and S. G. Lemay, Phys. Rev. Lett., 2004, 93, 170802 CrossRef CAS PubMed.
- M. Dishon, O. Zohar and U. Sivan, Langmuir, 2011, 27, 12977–12984 CrossRef CAS PubMed.
- J. E. Sader, J. W. M. Chon and P. Mulvaney, Rev. Sci. Instrum., 1999, 70, 3967–3969 CrossRef CAS.
- V. Valmacco, M. Elzbieciak-Wodka, C. Besnard, P. Maroni, G. Trefalt and M. Borkovec, Nanoscale Horiz., 2016, 1, 325–330 RSC.
- F. J. Montes Ruiz-Cabello, P. Maroni and M. Borkovec, J. Chem. Phys., 2013, 138, 234705 CrossRef PubMed.
- R. Pericet-Camara, G. Papastavrou, S. H. Behrens and M. Borkovec, J. Phys. Chem. B, 2004, 108, 19467–19475 CrossRef CAS.
- T. Oncsik, A. Desert, G. Trefalt, M. Borkovec and I. Szilagyi, Phys. Chem. Chem. Phys., 2016, 18, 7511–7520 RSC.
- A. Hakim, M. Nishiya and M. Kobayashi, Colloid Polym. Sci., 2016, 294, 1671–1678 CAS.
- H. D. Ackler, R. H. French and Y. M. Chiang, J. Colloid Interface Sci., 1996, 179, 460–469 CrossRef CAS.
- J. Luczak, J. Hupka, J. Thoming and C. Jungnickel, Colloids Surf., A, 2008, 329, 125–133 CrossRef CAS.
- V. Valmacco, M. Elzbieciak-Wodka, D. Herman, G. Trefalt, P. Maroni and M. Borkovec, J. Colloid Interface Sci., 2016, 472, 108–115 CrossRef CAS PubMed.
- J. G. Kirkwood and J. B. Shumaker, Proc. Natl. Acad. Sci. U. S. A., 1952, 38, 863–871 CrossRef CAS.
- N. Adzic and R. Podgornik, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 91, 022715 CrossRef PubMed.
- S. J. Miklavic, D. Y. C. Chan, L. R. White and T. W. Healy, J. Phys. Chem., 1994, 98, 9022–9032 CrossRef CAS.
- G. Silbert, D. Ben-Yaakov, Y. Dror, S. Perkin, N. Kampf and J. Klein, Phys. Rev. Lett., 2012, 109, 168305 CrossRef PubMed.
- I. Popa, G. Papastavrou and M. Borkovec, Phys. Chem. Chem. Phys., 2010, 12, 4863–4871 RSC.
- S. H. Donaldson, A. Royne, K. Kristiansen, M. V. Rapp, S. Das, M. A. Gebbie, D. W. Lee, P. Stock, M. Valtiner and J. Israelachvili, Langmuir, 2015, 31, 2051–2064 CrossRef CAS PubMed.
- R. F. Tabor, F. Grieser, R. R. Dagastine and D. Y. C. Chan, Phys. Chem. Chem. Phys., 2014, 16, 18065–18075 RSC.
- M. Kanduc, E. Schneck and R. R. Netz, Chem. Phys. Lett., 2014, 610, 375–380 CrossRef.
| This journal is © the Owner Societies 2018 |