
Colorimetric ELISA with an acid–base indicator for sensitive detection of ochratoxin A in corn samples†
Ke
Pei
ab,
Ying
Xiong
ab,
Xiangmin
Li
*b,
Hu
Jiang
b and
Yonghua
Xiong
 *ab
*ab
aState Key Laboratory of Food Science and Technology, Jiangxi-OAI Joint Research Institute, Nanchang University, 235 Nanjing East Road, Nanchang 330047, P. R. China. E-mail: yhxiongchen@163.com; Fax: +86-791-8833-3708; Tel: +86-791-8833-4578
bJiangxi-OAI Joint Research Institute, Nanchang University, 235 Nanjing East Road, Nanchang 330047, P. R. China. E-mail: lixiangmin73@163.com; Tel: +86-791-8833-3529
First published on 16th November 2017
Abstract
In this study, a direct competitive enzyme-linked immunosorbent assay (dcELISA) with an acid–base indicator was developed for the naked-eye detection of ochratoxin A (OTA). OTA-labeled urease conjugates were used as competing antigens, and the acid–base indicator of bromocresol purple (BCP) dye was employed as the signal reporter. When the competing antigen was captured by the anti-OTA antibody on the plate well, the urea substrate was catalyzed to generate NH3 molecules. The BCP dye produced a vivid color change from purple to light yellow because of a slight pH change in the solution, and hence, the indicator was suitable for the naked-eye detection of analytes. Under optimized conditions, the proposed method exhibited a high sensitivity for OTA naked-eye detection with a cutoff limit of 50 pg mL−1. The method also showed a good linear range of 12.5–100 pg mL−1 for OTA quantitative detection, with a half maximal inhibitory concentration of 31.4 pg mL−1, which is 11-fold lower than that of conventional HRP-based ELISA (IC50 = 354 pg mL−1). The reliability of the proposed colorimetric ELISA method for naked-eye detection indicated no false negative and only three false positive results from 70 tests. Intra- and inter-assays showed that the average recovery ranged from 83.7% to 111%, and the coefficient of variation (CV) ranged from 3.71% to 13.5%. The proposed method exhibited acceptable accuracy and precision for the quantitative detection of OTA in real corn samples. In summary, the developed acid–base indicator-based dcELISA offers a promising platform for resource-constrained countries for highly sensitive naked-eye detection of haptens.
1. Introduction
Ochratoxin A (OTA) is a common mycotoxin mainly generated by several Aspergillus and Penicillium species.1,2 OTA is characterized by nephrotoxicity, teratogenicity, and immunotoxicity and is thus categorized as a group 2B carcinogen by the International Agency for Research on Cancer.3–5 The European Commission has established regulatory levels for OTA in different foods such as dried fruits and soluble coffee (10 μg kg−1), raw cereal (5 μg kg−1), wine (2 μg kg−1), and baby food (0.5 μg kg−1).6,7 Various methods based on instrumental analysis have been developed for OTA determination; these methods include chromatography methods,8,9 infrared spectroscopy methods10,11 and capillary electrophoresis.12,13 These highly sensitive methods are widely used for OTA confirmation detection but minimally applied in resource-constrained regions because they require expensive and sophisticated instruments.14 One of the most popular methods for OTA screening is enzyme-linked immunosorbent assay (ELISA) through horseradish peroxidase (HRP) catalysis with tetramethylbenzidine as the signal reporter;15 this technique features low cost, simple operation, and a high throughput. However, the conventional competitive ELISA is not adaptable for naked-eye detection because it requires a plate reader to effectively differentiate the tonality of analytes with similar concentrations. Therefore, convenient and affordable methodologies for naked-eye detection of analytes must be developed particularly for resource-constrained countries.16,17Colorimetric ELISA with an acid–base indicator is an optimal method for the naked-eye determination of various harmful substances in resource-constrained regions. The principle of this acid–base indicator colorimetric ELISA is based on labels such as penicillinase,18 alkaline phosphatase19 or urease20 conjugate catalysis of their corresponding substrates to generate a slight pH change in solution. Subsequently, the acid–base indicator shows a sharp color change with respect to the pH change. In the present study, we chose OTA-labeled urease as the competing antigen for developing a colorimetric ELISA for OTA screening detection by the naked eye.
Over the past few decades, urease-based ELISA with a sandwich format has been widely used for the detection of various biological macromolecules, such as sex-specific antigens, snake venom, β-lactamase, plant viruses, blastomycosis, etc.21–25 Compared with HRP-based ELISA, urease-based sandwich ELISA shows several advantages.26 For example, urease-based sandwich ELISA allows for a sharp titration end point because of the vivid color change of bromocresol purple (BCP) (yellow to purple) with pH changes in the solution. Urease-based sandwich ELISA can also be used to detect cell-associated antigens and antibodies because the urease cannot occur in mammalian tissues. This method provides false positive results for the detection of complex samples because of the nonspecific adsorption of ammonia on sample matrices, such as proteins.27 In the present study, urease was first introduced as the carrier of a competing antigen to develop a direct competitive ELISA (dcELISA) for OTA detection. Owing to the strong hydrophobicity of OTA molecules, a solution containing a high concentration of methanol was necessary to ensure high extraction recovery from OTA-contaminated samples. The high concentration of methanol could denature the protein in the samples and eliminate the false positive result for the naked-eye detection of OTA. The specificity and cutoff limit of urease-based dcELISA for the qualitative naked-eye determination of OTA were also determined. In addition, the quantitative performance of the proposed method for OTA analysis was evaluated in terms of half maximal inhibitory concentration, accuracy, and precision for detection in real corn samples.
2. Experimental
2.1 Materials and apparatus
Ochratoxin A (OTA), deoxynivalenol (DON), zearalenone (ZEN), fumonisin B1 (FB1), aflatoxin B1 (AFB1), a 3,3′,5,5′-tetramethylbenzidine (TMB) liquid substrate system, N,N′-dicyclohexylcarbodiimide (DCC), N-hydroxysuccinimide (NHS), bovine serum albumin (BSA), protein G, urease (type III, from Canavalia ensiformis, 500–800 units per mL) and urea were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Mouse anti-OTA mAbs were obtained from Jiangxi Zodolabs Biotech Corp. (Jiangxi, China). Sodium hydroxide, sodium chloride, and bromocresol purple (BCP) were purchased from Shanghai Jingchun Scientifical Co., Ltd. Other reagents were of analytical grade and purchased from Sinopharm Chemical Corp (Shanghai, China). Chemicals and materials were used without further purification. Ultrapure water was obtained with a Milli-QA apparatus (Molsheim, France). The 96-well microplates (high binding, white) were obtained from Costar Inc. (Cambridge, MA, USA). The DNM-9602 microplate reader was purchased from Beijing Perlong New Technology Co., Ltd.2.2 Preparation of OTA labeled urease
OTA-labeled urease (urease@OTA) was synthesized by coupling the carboxyl group of OTA to the amino group of urease in the presence of DCC/NHS as described previously.28 Briefly, 0.1 mg of OTA was dissolved in 100 μL of anhydrous tetrahydrofuran and activated with NHS and DCC (the molar ratio of OTA, NHS and DCC was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:6) at room temperature under constant stirring for 90 min. The activated OTA solution was centrifuged at 10000g for 15 min to remove the precipitant. The supernatant was then added to the urease solution (0.01 mol L−1 bicarbonate buffer, pH 8.0) with the mole ratio of OTA/urease at 20:1, and reacted at room temperature for 3 h. The OTA@urease conjugates were dialyzed using 0.128 mol L−1 NaCl (pH 5.96) containing 20% (v/v) glycerinum to conserve its catalytic activity according to the user manual. The prepared OTA@urease conjugates were kept at 4 °C until further use.
:3:6) at room temperature under constant stirring for 90 min. The activated OTA solution was centrifuged at 10000g for 15 min to remove the precipitant. The supernatant was then added to the urease solution (0.01 mol L−1 bicarbonate buffer, pH 8.0) with the mole ratio of OTA/urease at 20:1, and reacted at room temperature for 3 h. The OTA@urease conjugates were dialyzed using 0.128 mol L−1 NaCl (pH 5.96) containing 20% (v/v) glycerinum to conserve its catalytic activity according to the user manual. The prepared OTA@urease conjugates were kept at 4 °C until further use.
2.3 Procedure of urease-based direct competitive ELISA
The 96-well microplates were first coated with 100 μL of protein G (20 μg mL−1) in bicarbonate buffer (100 mM, pH 8.6) at 4 °C overnight, and then blocked with 1 mg mL−1 BSA solution at 37 °C for 2 h. After washing three times with PBS buffer containing 0.05% Tween-20 (PBST, pH 7.4), 100 μL of anti-OTA mAbs (80.5 ng mL−1) was added and incubated at room temperature for 2 h. After washing with PBST three times, 50 μL of OTA@urease solution (1.94 μg mL−1) and 50 μL of the sample solution were added into each plate well. After incubation at 37 °C for 60 min, the microplates were washed thrice with 0.128 mol L−1 NaCl solution (pH 5.96) containing 0.05% Tween-20 and then further washed twice with pure water containing 0.128 mol L−1 NaCl solution (pH 5.96). Subsequently, 100 μL of the urea solution (dissolved 0.1 mol L−1 urea containing 0.128 mol L−1 NaCl solution, pH 5.96) was added to each well. After reaction at 37 °C for 30 min, 10 μL of the freshly prepared BCP solution (8 mg L−1) was added to each well.29 The vivid color change of BCP from purple to light yellow was used for OTA qualitative detection with the naked eye, while the OD588/434 value was used for the quantitative detection of OTA by using the microplate reader.2.4 Sample preparation
Real corn samples were collected from grain procurement agencies (Shandong, China) and stored in a freezer at −20 °C until further use. The sample extraction was performed according to our previous report.30 Briefly, the desired OTA concentration was added to the pulverized corn sample, which was confirmed to be OTA free by a LC-MS/MS method. The OTA spiked samples (1 g) were extracted by ultrasound treatment with 5 mL of 60% (v/v) methanol/water for 20 min. The mixture was centrifuged at 12000g for 15 min. The resulting suspension was diluted by adding 0.128 mol L−1 NaCl (pH 5.96) to obtain a final methanol concentration of 10% (v/v) for the quantitative or qualitative detection of OTA.
3. Results and discussion
3.1 Principle of urease-based dcELISA for OTA determination
In the present dcELISA, the HRP was altered by urease, and TMB was replaced with the BCP molecule. BCP is a kind of pH-sensitive dye, and its tonality shows a vivid change from bright yellow to purple as the pH increases from 5.8 to 7.5.31,32 The detailed schematic of urease-based dcELISA is shown in Fig. 1. When the OTA molecule was absent in the sample solution, urease@OTA conjugates (competing antigens) were captured by anti-OTA mAbs in the microplate. Urease catalyzed urea to generate NH3; as a result, the acid–base indicator (BCP molecule) would produce a purple color because of the increase in the pH of the solution. In contrast, when the OTA molecule is present in the sample solution, the OTA would competitively bind to anti-OTA mAbs in the microplate, resulting in a less amount of urea being hydrolyzed by urease. Thus, the BCP exhibited a light yellow color because of a negligible pH change in the solution. Therefore, the sharp color change from purple to light yellow can facilitate the detection of target analytes with the naked eye. | ||
| Fig. 1 Schematic of acid–base indicator colorimetric ELISA for ochratoxin A detection. | ||
3.2 Development of urease-based dcELISA
At different pH levels, the BCP molecule has two kinds of degenerate intramolecular charge transfer states.33 In acidic solution, most of the BCP molecules lose their electrons and obtain a positive charge; thus, the color of the BCP solution appears light yellow. When the solution pH increases, the BCP molecules with negative charge increase and the BCP color changes from light yellow to purple. The UV-vis absorption spectra of the BCP solution at different pH levels are shown in Fig. 2A. The maximum absorption peak of most BCP molecules is 434 nm when the solution pH is 5.96 and red shifts to 588 nm when the solution pH increases to 6.53. The accurate pH transition points of the BCP dye were optimized to improve the sensitivity of the proposed method. First, we prepared a series of solutions with pH values varying from 5.96 to 6.53 by adding different ratios of 0.5 mmol L−1 NaOH solutions to pure water. The results in Fig. 2B indicated that when the solution pH is 5.96, the BCP solution produces a light yellow color with a very low OD588/434 value of 0.249; when the solution pH slightly increases to 6.25, the BCP dye shows an obvious change of color from light yellow to brownish red with the OD588/434 value rising to 0.575; when the pH value further increases to 6.38, the BCP solution produces a vivid color change to purple, while the OD588/434 value sharply increases to 1.116. These results indicated that the BCP dye exhibits a sensitive optical behavior corresponding to a very narrow pH change from 5.96 to 6.38 (ΔpH < 0.4). Thus, the pH for urease catalysis of urea in the immunoassay should be adjusted to 5.96 to improve the sensitivity of the proposed method. The effects of urea and urease@OTA on BCP color changes in pH 5.96 NaCl solution were evaluated. The results in Fig. S1† showed that the addition of 0.1 mol L−1 urea or 100 μg mL−1 urease@OTA cannot change the BCP color in 0.128 mol L−1 NaCl solution (pH 5.96). By contrast, the addition of urease@OTA to the mixture of urea (0.1 mol L−1) containing 0.128 mol L−1 NaCl solution induced a vivid color change in the BCP solution from light yellow to purple after 30 min of incubation at 37 °C. Hence, urease or urea alone at a certain concentration could not influence changes in the BCP color.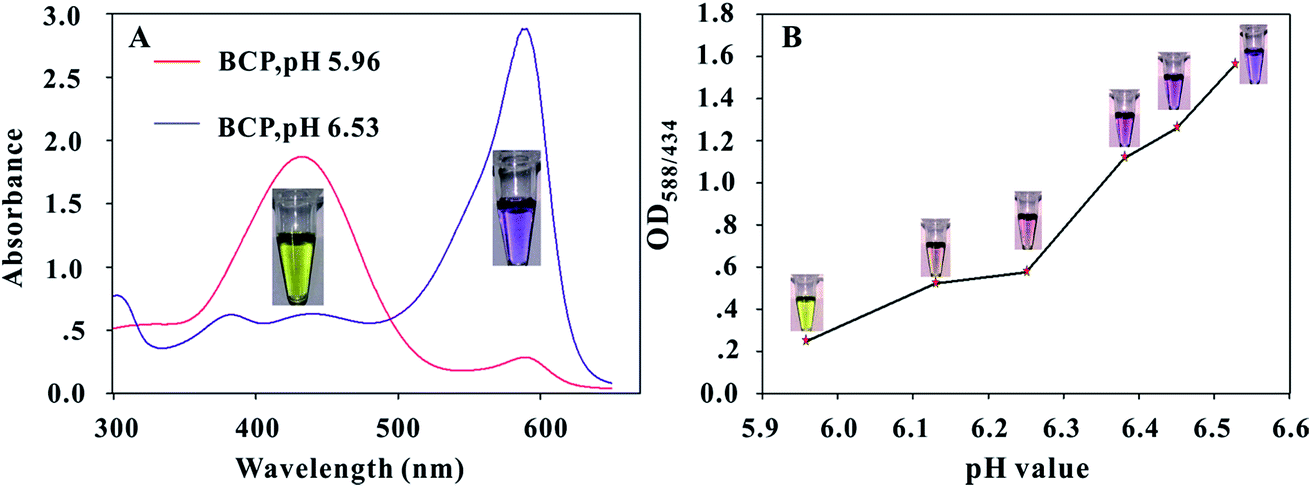 | ||
| Fig. 2 Characterization of the pH transition points of BCP dye at different pH levels. (A) UV-vis absorption spectra of the BCP solution with pH values at 5.96 and 6.53, respectively; (B) changes in the color and OD588/434 values of BCP dye at different pH levels from 5.96 to 6.53. | ||
The urease@OTA conjugate (competing antigen), an important component in urease-based dcELISA, was synthesized using an activated ester method. The UV-vis spectra of OTA@urease, urease, and OTA are shown in Fig. S2.† The urease solution exhibits a protein characteristic peak at 280 nm, whereas the OTA solution shows an absorption peak at 320 nm. The urease@OTA conjugates show two absorption peaks at 280 and 363 nm. The red-shifted absorption peak at 363 nm indicates that the OTA molecule has been successfully coupled with the urease protein. The coupled molar ratio of OTA@urease is calculated to be 5.73:1 (the detailed description is shown in the ESI†). To improve the bioactivity of anti-OTA mAbs, the 96-well microplates were pre-coated with protein G to directly capture the Fc fragment of mAbs. The concentrations of anti-OTA mAbs and the urease@OTA were optimized by the chessboard titration method. The detailed results are shown in Table S1,† indicating that the optimal concentrations of anti-OTA mAbs and urease@OTA were 80.5 ng mL−1 and 1.94 μg mL−1, respectively, while the BCP solution obviously produced a purple color with an OD588/434 value of 1.402.
In the immunoassay, pH change could influence anti-OTA mAb and urease@OTA immunoreactions. Meanwhile, the solubility of the OTA molecule in acidic or neutral aqueous solution is very low. An extraction solution containing 60% (v/v) methanol is required to obtain a high extraction recovery from OTA-contaminated food samples. However, high methanol concentration in the solution can disturb the antigen–antibody reaction. Thus, the effects of pH and methanol concentration on the sensitivity of dcELISA were investigated. The results in Fig. 3A showed that the OD588/434 value increased sharply from 1.26 to 1.57 with increasing solution pH value from 5.0 to 6.0, and then gradually decreased with the pH value further increasing to 9.0. Thus, the optimum pH value for urease@OTA and anti-OTA mAb interaction is 6.0. The results in Fig. 3B indicated that the maximum value of OD588/434 was obtained to be 1.59 when the methanol concentration was 10%. Considering the high sensitivity of the urease-based dcELISA, the actual sample extract was suggested to be diluted with 0.128 mol L−1 NaCl solution (pH 5.96) to a final methanol concentration of 10%. In addition, the urea concentration, the incubation time, and the temperature of urease catalysis of urea are also key factors that influence the BCP color change and the sensitivity of urease-based dcELISA. The results in Fig. 3C showed that the OD588/434 value sharply increased with increasing urea concentration, and then achieved a maximum of 1.6 when the urea concentration was 0.1 mol L−1; when the urea concentration further increased to 1.0 mol L−1, the OD588/434 value showed a slight decline to 1.43, indicating that excessive urea concentration could inhibit urease activity. The optimal temperature of hydrolysis of urea by urease is 60 °C according to a previous report. However, the results in Fig. 3D showed that the maximum OD588/434 value of 1.61 was achieved at 37 °C. Presumably, a higher catalytic temperature such as 45 °C could cause ammonia volatilization from the well, and thus will result in a low OD588/434 value. Fig. 3E reveals that the OD588/434 value gradually increased with enzyme catalysis time extending, and then reached a plateau when the catalysis time was 30 min. Based on the above results, the optimized experimental conditions for urease-based dcELISA were as follows: the immunoreaction between mAbs and urease@OTA was performed in 0.128 mol L−1 NaCl solution (pH 5.96) containing 10% (v/v) methanol; urease catalysis of urea was carried out at 37 °C for 30 min with 0.1 mol L−1 urea concentration.
 | ||
| Fig. 3 Optimization of the experimental conditions of urease-based dcELISA. Effects of different pH values (A) and methanol concentrations (B) on the immunoreaction between anti-OTA mAbs and urease@OTA; effects of different urea concentrations (C), incubation time (D) and temperature (E) on the urease catalysis of urea in urease-based dcELISA. The vertical bars indicate the standard deviation (n = 3). | ||
3.3 Analytical performance of the modified-ELISA
OTA standard solutions were prepared by diluting the OTA stock solution with 0.128 mol L−1 NaCl solution containing 10% methanol to a final OTA concentration from 0 pg mL−1 (as negative control) to 1600 pg mL−1. Under optimized conditions, each OTA standard solution was investigated by the urease-based dcELISA. As shown in the inset of Fig. 4A, the BCP solution would produce a vivid color change from purple to light yellow (or gray) with OTA concentration increasing from 0 to 50 pg mL−1; the bright yellow (gray) with purple tonality is easily distinguishable by the naked eye. Thus, an OTA concentration of 50 pg mL−1 was defined as the cutoff limit of the proposed method for OTA qualitative analysis by the naked eye. The OTA quantitative calibration curve was constructed by plotting the B/B0 ratios against the logarithm of various OTA concentrations, where B0 and B denote the OD588/434 value of the negative control and the positive samples, respectively. Fig. 4A also shows the calibration curve of the developed dcELISA, which exhibited a good linear range from 12.5 pg mL−1 to 100 pg mL−1 with a reliable correlation coefficient (R2 = 0.99). The regression equation could be represented as y = −0.3391ln(x) + 1.7688, where y is the B/B0, and x is the logarithm of OTA concentration. Error bars were based on three duplicate measurements at different OTA concentrations. The IC50 value was calculated to be 31.4 pg mL−1, which is 11-fold lower than that of conventional HRP-based ELISA (IC50 = 354 pg mL−1; Fig. S3†). The specificity of the developed method was determined by analyzing an OTA solution (100 pg mL−1) and four other common mycotoxin solutions (1 ng mL−1), including ZEN, DON, FB1, and AFB1. A negative control test was performed by adding 0.128 mol L−1 NaCl solution (pH 6.0, containing 10% methanol). Fig. 4B shows that the changes in the OD588/434 values of ZEN, DON, FB1, AFB1 and OTA negative samples were negligible, whereas the OD588/434 value for the OTA sample was significantly less than 0.4. These results demonstrated the excellent selectivity of the developed urease-based dcELISA for OTA determination.
 | ||
| Fig. 4 (A) Quantitative calibration curve of OTA determination using the urease-based dcELISA. The spiked standard solution in 0.128 M NaCl (pH 5.96) solution containing 10% methanol with OTA concentrations ranging from 0 pg mL−1 (as negative control) to 1600 pg mL−1. Inset figure: linear plots of B/B0 as a function of the logarithm of OTA concentration. The error bars represent the standard deviation of three independent measurements (n = 3). (B) Specificity experiments for OTA, ZEN, DON, FB1 and AFB1. (1) Control, (2) AFB1 (1 ng mL−1), (3) FB1 (1 ng mL−1), (4) ZEN (1 ng mL−1), (5) DON (1 ng mL−1), (6) AFB1 (1 ng mL−1) + ZEN (1 ng mL−1) + DON (1 ng mL−1) + FB1 (1 ng mL−1), (7) OTA (100 pg mL−1), and (8) OTA (100 pg mL−1) + AFB1 (1 ng mL−1) + ZEN (1 ng mL−1) + DON (1 ng mL−1) + FB1 (1 ng mL−1). The vertical bars indicate the standard deviation (n = 3). A negative control test was performed by adding 0.128 M mol L−1 NaCl (pH 5.96) containing 10% methanol. | ||
The reliability of the proposed method for OTA qualitative detection with the naked eye was evaluated by analyzing OTA-spiked samples in corn extracts, which were prepared by adding OTA stock solution to OTA-free diluted corn solution at OTA concentrations of 0, 10, 20, 50, 60, 100, and 160 pg mL−1. The diluted corn solution was obtained by diluting the corn extract with 0.128 mol L−1 NaCl solution to a final methanol concentration of 10%. All samples were analyzed 10 times using the proposed method. The results in Table 1 showed no false negative with three false positive results in the 70 tests, demonstrating the acceptable method accuracy for OTA qualitative analysis with the naked eye (the corresponding stereogram is shown in Fig. S4†). To further evaluate the practicability of the proposed dcELISA for OTA quantitative detection in actual samples, four OTA-free corn samples were fortified with five different OTA concentrations of 1 μg kg−1, 5 μg kg−1, 20 μg kg−1, 40 μg kg−1, and 60 μg kg−1. The intra- and inter-assays were performed with three replicates at each spiked concentration. The intra-assay was carried out within one day, and the inter-assay was completed with three replicates. Table 2 shows that the average recoveries of the intra-assay ranged from 83.7% to 108.8% with a coefficient of variation (CV) ranging from 3.71% to 13.5%, and those for the inter-assay ranged from 86.2% to 111% with the CV value ranging from 5.10% to 13.4%. The above results demonstrate the acceptable accuracy and precision of urease-based dcELISA for the quantitative analysis of OTA in real corn samples.
| OTA spiked concentration (pg mL−1) | Number of samples | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| a Notes: +, yellow or gray color in the well, OTA-positive results; −, purple color in the well, OTA-negative results. | ||||||||||
| 0 | − | − | − | − | − | − | − | − | − | − |
| 10 | − | − | − | − | − | − | − | − | − | + |
| 20 | − | + | − | − | − | + | − | − | − | − |
| 50 | + | + | + | + | + | + | + | + | + | + |
| 60 | + | + | + | + | + | + | + | + | + | + |
| 100 | + | + | + | + | + | + | + | + | + | + |
| 160 | + | + | + | + | + | + | + | + | + | + |
| OTA added (μg kg−1) | Intra-assay (n = 3)a | Inter-assay (n = 3)b | ||||
|---|---|---|---|---|---|---|
| Mean ± SD (μg kg−1) | Recovery (%) | CV (%) | Mean ± SD (μg kg−1) | Recovery (%) | CV (%) | |
| a The assay was carried out in triplicate on the same day. b The assay was performed on three consecutive days. | ||||||
| 1.00 | 0.868 ± 0.074 | 86.8 | 8.52 | 1.11 ± 0.135 | 111 | 12.2 |
| 5.00 | 4.75 ± 0.176 | 95.0 | 3.71 | 4.31 ± 0.437 | 86.2 | 10.1 |
| 20.0 | 19.78 ± 2.14 | 98.9 | 10.8 | 19.9 ± 1.40 | 99.5 | 7.04 |
| 40.0 | 43.5 ± 5.10 | 108.8 | 11.7 | 39.2 ± 2.00 | 98.0 | 5.10 |
| 60.0 | 50.2 ± 6.79 | 83.7 | 13.5 | 55.5 ± 7.43 | 92.5 | 13.4 |
4. Conclusions
We presented an acid–base indicator-based dcELISA for OTA detection using urease and OTA conjugates as competing antigens. The proposed ELISA exhibited a very high sensitivity for the naked-eye determination of OTA with a cutoff limit of 50 pg mL−1. With the aid of a microplate reader, the proposed method also showed a dynamic linear detection range from 12.5 pg mL−1 to 100 pg mL−1 with the IC50 value at 31.4 pg mL−1, which is approximately 11-fold lower than that of conventional HRP-based ELISA. In addition, the proposed method also showed an acceptable accuracy for OTA qualitative or quantitative determination. Notably, the washing step of the proposed ELISA method is crucial for experimental accuracy and repeatability because the BCP dye is extremely sensitive to pH change in the solution. In summary, the proposed method provides a new tool for the sensitive detection of small chemical pollutants by the naked eye in resource-constrained countries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the National Basic Research Program of China (2013CB127804), Training Plan for the Main Subject of Academic Leaders of Jiangxi Province (20142BCB22004), and the Open Project Program of State Key Laboratory of Food Science and Technology of Nanchang University (No. SKLF-KF-201609).References
- C. Zaied, S. Abid, L. Zorgui, C. Bouaziz, S. Chouchane, M. Jomaa and H. Bacha, Food Control, 2009, 20, 218–222 CrossRef CAS.
- S. Ozden, A. S. Akdeniz and B. Alpertunga, Food Control, 2012, 25, 69–74 CrossRef CAS.
- T. Kuiper-Goodman, Food Addit. Contam., 1996, 13, 53–57 CrossRef CAS PubMed.
- B. Kabak and A. D. Dobson, Crit. Rev. Food Sci. Nutr., 2017, 57, 18–34 CAS.
- W. H. Organization and I. A. f. R. O. Cancer, IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, 1993, p. 56 Search PubMed.
- European Commission, Commission Regulation (EC) No. 472/2002 of 12 March 2002 amending Regulation (EC) No. 466/2001, Off. J. Eur. Communities: Legis., 2002, 75, 18–20 Search PubMed.
- European Commission, Commission Regulation (EC) No. 123/2005 of 26 January 2005 amending Regulation (EC) No. 466/2001, Off. J. Eur. Communities: Legis., 2005, 25, 3–5 Search PubMed.
- L. Marino-Repizo, F. Kero, V. Vandell, A. Senior, M. Isabel Sanz-Ferramola, S. Cerutti and J. Raba, Food Chem., 2015, 172, 663–668 CrossRef CAS PubMed.
- T. Rodriguez-Cabo, I. Rodriguez, M. Ramil and R. Cela, Food Chem., 2016, 199, 401–408 CrossRef CAS PubMed.
- P. Taradolsirithitikul, P. Sirisomboon and C. Dachoupakan Sirisomboon, J. Sci. Food Agric., 2017, 97, 1260–1266 CrossRef CAS PubMed.
- A. C. Galvis-Sánchez, A. Barros and I. Delgadillo, Food Addit. Contam., 2007, 24, 1299–1305 CrossRef PubMed.
- S. Almeda, L. Arce and M. Valcárcel, Electrophoresis, 2008, 29, 1573–1581 CrossRef CAS PubMed.
- N. Arroyo-Manzanares, L. Gámiz-Gracia and A. M. García-Campaña, Food Chem., 2012, 135, 368–372 CrossRef CAS PubMed.
- J. H. Soh, Y. Lin, S. Rana, J. Y. Ying and M. M. Stevens, Anal. Chem., 2015, 87, 7644–7652 CrossRef CAS PubMed.
- P. D. Josephy, T. Eling and R. P. Mason, J. Biol. Chem., 1982, 257, 3669–3675 CAS.
- J. C. Patton, A. H. Coovadia, T. M. Meyers and G. G. Sherman, Clin. Vaccine Immunol., 2008, 15, 388–391 CrossRef CAS PubMed.
- S. Tang and I. Hewlett, J. Infect. Dis., 2010, 201(suppl. 1), S59–S64 CrossRef CAS PubMed.
- S. A. Wada, Y. Matsui, T. Tsuda and M. Wade, J.– Assoc. Off. Anal. Chem., 1988, 71(2), 373–374 Search PubMed.
- C. Mousty, O. Kaftan, V. Prevot and C. Forano, Sens. Actuators, B, 2008, 133, 442–448 CrossRef CAS.
- H. H. Deng, G. L. Hong, F. L. Lin, A. L. Liu, X. H. Xia and W. Chen, Anal. Chim. Acta, 2016, 915, 74–80 CrossRef CAS PubMed.
- V. Achal and X. Pan, Curr. Microbiol., 2011, 62, 894–902 CrossRef CAS PubMed.
- H. M. Chandler and J. G. Hurrell, Clin. Chim. Acta, 1982, 121, 225–230 CrossRef CAS.
- G. Scalarone, J. Med. Vet. Mycol., 1987, 25, 215–222 CrossRef CAS PubMed.
- R. Premier, J. Cox, D. Aitken and K. Healey, J. Immunol. Methods, 1985, 83, 371–377 CrossRef CAS PubMed.
- M. C. Cerrone and R. E. Kuhn, J. Immunol. Methods, 1991, 138, 65–75 CrossRef CAS PubMed.
- H. Chandler, J. Cox, K. Healey, A. MacGregor, R. Premier and J. Hurrell, J. Immunol. Methods, 1982, 53, 187–194 CrossRef CAS PubMed.
- A. Abraham and S. Albrechtsen, Z. Pflanzenkrankh. Pflanzenschutz, 2001, 49–57 CAS.
- L. Yu, Y. Zhang, C. Hu, H. Wu, Y. Yang, C. Huang and N. Jia, Food Chem., 2015, 176, 22–26 CrossRef CAS PubMed.
- H. Shi, X. Chen, L. Li, L. Tan, X. Ren, J. Ren and X. Meng, Dalton Trans., 2014, 43, 9016–9021 RSC.
- H. Duan, X. Chen, W. Xu, J. Fu, Y. Xiong and A. Wang, Talanta, 2015, 132, 126–131 CrossRef CAS PubMed.
- Y. Lvov, A. A. Antipov, A. Mamedov, H. Möhwald and G. B. Sukhorukov, Nano Lett., 2001, 1, 125–128 CrossRef CAS.
- L. J. Schussel and J. E. Atwater, Chemosphere, 1995, 30, 985–994 CrossRef CAS.
- C. J. Talone, J. Gao, J. R. Lynch, R. M. Tanu and S. T. Deyrup, Spectrochim. Acta, Part A, 2016, 156, 138–142 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ay01959a |
| This journal is © The Royal Society of Chemistry 2018 |