
Cell to rodent: toxicological profiling of folate grafted thiomer enveloped nanoliposomes
 c
Gul
Shahnaz
*a
c
Gul
Shahnaz
*a
Abstract
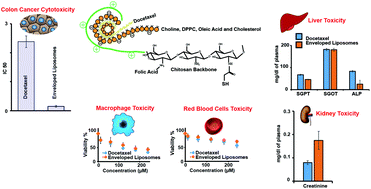
Polymeric nanomaterials, hybridized with lipid components, e.g. phosphocholine or fatty acids, are currently being explored for efficient nano-platforms for hydrophobic drugs. However, their toxicology and toxicokinetics need to be established before enabling their clinical potential. The aim of this study was to investigate the toxicological profile of thiomer enveloped hybrid nanoliposomes (ENLs) and bare nanoliposomes (NLs), loaded with docetaxel (DTX) hydrophobic drug, biocompatible nano-carriers for therapeutic cargo. The in vitro toxicity of hybrid ENLs and NLs was evaluated towards the HCT-116 colon cancer cell line. Biocompatibility was explored against macrophages and acute oral toxicity was examined in mice for 14 days. The anticancer IC50 for ENLs was 0.148 μg ml−1 compared with 2.38 μg ml−1 for pure docetaxel (DTX). The human macrophage viability remained above 65% and demonstrated a high level of biocompatibility and safety of ENLs. In vivo acute oral toxicity showed slight changes in serum biochemistry and haematology but no significant toxicities were observed referring to the safety of DTX loaded hybrid ENLs. On histological examination, no lesions were determined on the liver, heart and kidney. These studies showed that hybrid ENLs can serve as a safe and biocompatible platform for oral delivery of hydrophobic drugs.
 Please wait while we load your content...
Please wait while we load your content...