DOI:
10.1039/C7TC02992F
(Paper)
J. Mater. Chem. C, 2017,
5, 10761-10769
K2MnF6 as a precursor for saturated red fluoride phosphors: the struggle for structural stability
Received
4th July 2017
, Accepted 13th September 2017
First published on 9th October 2017
Abstract
Phosphor-converted white light-emitting diodes (LEDs) are currently taking over the lighting market because of their high luminous efficiency, environmentally friendly nature and long lifetime. A new generation of saturated red fluoride phosphors, using Mn4+ as the activator, has gained interest in further enhancing the color rendering properties and efficiency of white LEDs for lighting and display applications. They can be described as A2MF6:Mn4+ (A = K, Na, Sc, NH4 and M = Si, Ge, Ti, Sn), KNaMF6:Mn4+, BaMF6:Mn4+ (M = Si, Ti) or ZnMF6·H2O (M = Si, Ge) compounds, in which Mn is a substitute for the M(IV) element of the fluoride host. A two-step co-precipitation synthesis method has recently been developed because of the increased control of the Mn valence state and the relatively low cost. In this method, K2MnF6 is first synthesized as a precursor which then serves as a source for the preparation of [MnF6]2− complexes in further phosphor synthesis. In-house production of K2MnF6 is required as it quickly degrades. Here, we investigate the structural properties after synthesis, as well as the main degradation routes of K2MnF6 when the material is subjected to heat and humidity or used in further synthesis reactions. It is found that impurities, such as KHF2, K2MnF5·H2O and Mn ions in an oxygen coordination, can be formed as a result of parasitic reactions during synthesis. Even in pure K2MnF6, degradation occurs due to heat and hydrolysis both of which induce reduction of the Mn4+ ion. Heating in air causes the material to form Mn2+ as KMnF3/KF·MnF2 starts to form at high temperatures due to hydrolysis. In dilute HF solutions the Mn4+ ion is partially reduced to Mn3+, often incorporated in hydrated structures as KMnF4·H2O and K2MnF5·H2O. The Mn3+ ion is found to affect the optical absorption properties.
Introduction
Ever since the introduction of high-brightness blue light-emitting diodes (LEDs), much effort has been put into the development of white LEDs (wLEDs). Progress in the field of lighting has come to the point at which wLEDs are now taking over the market due to their improved stability and efficiency in comparison with more traditional light sources such as incandescent or fluorescent lamps.1–3 wLEDs can be based on the combination of three separate LED chips which emit blue, green and red light, respectively. However most wLEDs make use of at least one photoluminescent material, a phosphor, hence the name phosphor-converted LEDs (pc-LEDs).4 Here, the combined emission from the blue LED chip and the phosphors yields white light. Y3Al5O12:Ce3+ (YAG:Ce) is often used in single-phosphor pc-LEDs because of the high stability and efficiency of this phosphor.2,5 However, without any additional phosphors, the obtained white light lacks in the green and red parts of the optical spectrum, hence leading to a high correlated color temperature (CCT) and a poor color rendering index (CRI) (CCT > 6000 K, Ra < 80).6 Therefore, other phosphor materials are often used in a phosphor blend. By adding a red component, one can lower the color temperature of wLEDs yielding warm-white light. On the one hand, the red phosphor emission above 650 nm should be minimal, as the human eye sensitivity is very low in this wavelength region. On the other hand, especially for display applications, saturated red phosphors are preferred as these enlarge the color gamut of the display (e.g. BT. 2020 guidelines for UHD displays7). Suitable new red phosphor materials should therefore show a saturated red emission below 650 nm.8 Apart from the optical requirements, the stability of red phosphor materials should also be adressed. Degradation of phosphor materials can lead to color changes or lumen losses over time when the material is stressed by heat (with temperatures up to 200 °C in high brightness LEDs9) or humidity from the environment. The study on the prevention of degradation becomes increasingly more important as the lifetime of current LEDs is expected to be at least 50.000 hours. In the past, sulfides and especially nitrides, e.g. (Sr,Ca)S:Eu2+,10 CaAlSiN3:Eu2+,11 and (Sr,Ba)2Si5N8:Eu2+,12 have been commercially used as red phosphor materials.
However, drawbacks such as poor thermal quenching and hydrolysis of the sulfides and a too broad emission band for many nitride phosphors, led to research towards a new generation of saturated red-emitting phosphors which do not involve rare-earth dopants.8 These phosphors, using Mn4+ as an activator13 in a fluoride host, are promising as they show saturated red emission below 650 nm and low thermal quenching at LED operating temperatures. K2SiF6:Mn4+, with an emission peak around 630 nm, can be considered as a prototype material for Mn4+ fluorides.14 Its synthesis and properties have been recently reviewed.15 BaSiF6:Mn4+,16 KNaSiF6:Mn4+,17 K2TiF6:Mn4+,18,19 Cs2TiF6:Mn4+,20 K2GeF6:Mn4+,21 Na2GeF6:Mn4+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 and BaGeF6:Mn4+23 are just a few examples of fluoride phosphors with a similar emission spectrum which can be attributed to Mn4+ and its fluorine ligands. BaMF6:Mn4+ (M = Si, Ge), KNaMF6:M4+ and A2MF6:Mn4+ (A = K, Na, Sc, NH4 and M = Si, Ge, Ti, Sn) red phosphors have Mn4+ surrounded by six fluorine atoms, creating [MnF6]2− complexes with an octahedral shape. These complexes show a characteristic saturated red emission around 630 nm upon blue or UV excitation, which is ideal for LED applications. While solid state reactions are often used to produce conventional phosphors, wet chemical synthesis routes are used for fluoride phosphors. The main challenge during the synthesis proves to be the large variety of valence states that Mn can adopt, ranging from 0 to 7+. In the past, synthesis of fluoride phosphors has been performed using various methods including wet chemical etching,17,21,22,24,25 redox precipitation14,26 and co-crystallization (cation exchange).19,27,28 Furthermore, hydrothermal methods16,19,23 have also been proposed. More recently, an improved two-step co-precipitation method has gained strong interest because of the improved control of the Mn valence.16,18,20,29 K2MnF6 is synthesized during the first synthesis step.18,28,30–32 This precursor material then provides anionic [MnF6]2− complexes, which can be incorporated into various fluoride host materials such as K2TiF6 or K2SiF6. Fluorine coordination of Mn stabilizes the 4+ valence state, preventing reduction towards lower valence states. Nevertheless, K2MnF6 is not commercially available due to its instability with respect to heat and moisture, requiring a delicate in-house synthesis. As defects and impurities in the final phosphors, caused by impure or degraded K2MnF6, can affect thermal quenching and phosphor efficiency,9,33 a thorough understanding of the synthesis and degradation of K2MnF6 is in place. Here, we investigate the structural and optical properties of K2MnF6 after synthesis as well as the effect of heat, humidity and further phosphor synthesis steps.
22 and BaGeF6:Mn4+23 are just a few examples of fluoride phosphors with a similar emission spectrum which can be attributed to Mn4+ and its fluorine ligands. BaMF6:Mn4+ (M = Si, Ge), KNaMF6:M4+ and A2MF6:Mn4+ (A = K, Na, Sc, NH4 and M = Si, Ge, Ti, Sn) red phosphors have Mn4+ surrounded by six fluorine atoms, creating [MnF6]2− complexes with an octahedral shape. These complexes show a characteristic saturated red emission around 630 nm upon blue or UV excitation, which is ideal for LED applications. While solid state reactions are often used to produce conventional phosphors, wet chemical synthesis routes are used for fluoride phosphors. The main challenge during the synthesis proves to be the large variety of valence states that Mn can adopt, ranging from 0 to 7+. In the past, synthesis of fluoride phosphors has been performed using various methods including wet chemical etching,17,21,22,24,25 redox precipitation14,26 and co-crystallization (cation exchange).19,27,28 Furthermore, hydrothermal methods16,19,23 have also been proposed. More recently, an improved two-step co-precipitation method has gained strong interest because of the improved control of the Mn valence.16,18,20,29 K2MnF6 is synthesized during the first synthesis step.18,28,30–32 This precursor material then provides anionic [MnF6]2− complexes, which can be incorporated into various fluoride host materials such as K2TiF6 or K2SiF6. Fluorine coordination of Mn stabilizes the 4+ valence state, preventing reduction towards lower valence states. Nevertheless, K2MnF6 is not commercially available due to its instability with respect to heat and moisture, requiring a delicate in-house synthesis. As defects and impurities in the final phosphors, caused by impure or degraded K2MnF6, can affect thermal quenching and phosphor efficiency,9,33 a thorough understanding of the synthesis and degradation of K2MnF6 is in place. Here, we investigate the structural and optical properties of K2MnF6 after synthesis as well as the effect of heat, humidity and further phosphor synthesis steps.
Experimental
K2MnF6 powders were prepared using the method of Bode and Bandte.32 Here, 36 g of KF and 2 g of KMnO4 were dissolved in 150 ml of 40% HF, preserving a 1:49 – KMnO4:KF ratio. The excess KF facilitates fluorine complexation of the Mn ions while the acidic synthesis environment limits hydroxide formation during synthesis. Reduction of manganese from Mn7+ to Mn4+ was performed by dropwise addition of 35% H2O2 with vigorous stirring. The addition rate of the reducing agent was controlled to prevent parasitic hydrolysis reactions, exclude excessive reduction of the Mn ions and control the formation of O2. Adding H2O2 induces a colour change of the solution which turns from purple, over light purple towards dark and finally light brown. Furthermore, a yellow precipitate forms on the bottom of the reaction cup. Stopping the H2O2 addition at the right moment is crucial but rather challenging due to ongoing reactions even after the last addition of H2O2. However, one typically stops the addition as soon as the solution evolves to light brown. For the aforementioned synthesis conditions, addition of around 2 ml of H2O2 proved to yield the maximal amount of K2MnF6 while minimizing the amount of impurity phases. After decantation, filtration and a washing treatment with HF and acetone, the precipitate can be dried and a bright yellow K2MnF6 powder sample is obtained. The overall synthesis reaction can be described as:31
| 2KMnO4 + 2KF + 10HF + 3H2O2 → 2K2MnF6 + 8H2O + 3O2↑ |
Structural properties were investigated by in- and ex situ X-ray diffraction (XRD) experiments. During in situ XRD measurements, a sample was placed on a heating stage in a reaction chamber filled with air or N2 and subsequently heated at a rate of 0.2 °C s−1 from room temperature (RT) up to 600 °C. A Bruker Advance D8 diffractometer equipped with a linear X-ray detector was used for in situ XRD monitoring of the samples in a 2θ range of 30.6° to 49.6°. Ex situ measurements in air were performed using both a Bruker Advance D8 and a Bruker D5000 diffractometer. Cu Kα1 tube emission was used in all XRD measurements. A Memmert HCP 108 weathering chamber was used to maintain ambient atmospheres with predefined heat and humidity levels in which samples could be aged over time. Optical diffuse reflectance measurements on the powders were performed using a Varian Cary 500 spectrometer, equipped with an internal 110 mm BaSO4-coated integrating sphere. X-ray absorption spectroscopy (XAS) at the Mn K edge was performed at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France, beam line BM26A.
Results and discussion
As previously mentioned, manganese can take on many valence states, the most common ones being 0, 2+, 3+, 4+ and 7+. This creates a challenge for the synthesis because Mn ions are sensitive to valence changes and in particular excessive reduction if the temperature and/or H2O2 addition rate are insufficiently controlled. Moreover, in the following, it will be shown that parasitic species often exist after the synthesis of K2MnF6, potentially contaminating further phosphor synthesis steps and affecting both structural and luminescence properties of the final phosphor. While some impurities can be eliminated by a washing treatment, other impurities can remain present in K2MnF6, often without any trace in the X-ray diffraction pattern. Even when purified, care should be taken as any additional reaction step might cause K2MnF6, and hence the [MnF6]2− complexes, to degrade.
Structural properties
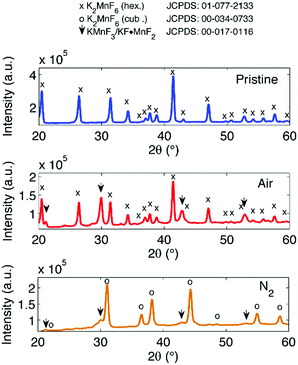
K2MnF6 exhibits two crystalline phases: a low-temperature hexagonal phase and a high-temperature cubic phase. Both structures are displayed in Fig. 1. MnF6 octahedrons are clearly visible in both phases. However, in the hexagonal phase, two distinct Mn sites are present which exhibit a different orientation of the [MnF6]2− coordination complex. Half of the complexes are tilted around the a-axis. The complexes in the cubic phase only have one orientation making all Mn sites identical. Phase transition from the hexagonal to the cubic structure occurs around 440 °C in an inert N2 atmosphere as can be seen in the in situ XRD pattern shown in the upper part of Fig. 2. Here, the XRD pattern within a 2θ range of 30.6° to 49.6° is shown as a function of increasing sample temperature, on going from RT up to 500 °C. Heating was performed under an inert N2 atmosphere to prevent any moisture-induced degradation of K2MnF6. The RT ex situ XRD patterns of the pristine hexagonal and cubic phase after heating in N2 are shown in Fig. 3. The final cubic structure could be maintained when the sample was cooled to room temperature. However, after storage of the powder in air for 17 days, the cubic phase almost completely returned to the hexagonal structure indicating that the cubic phase is not stable over time at room temperature.
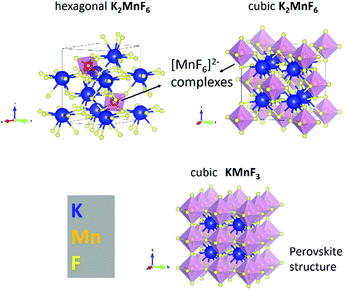 |
| | Fig. 1 Top: Unit cell of the hexagonal (low temperature) and cubic (high temperature) phase of K2MnF6. The purple semi-transparent octahedrons represent the [MnF6]2− complexes. Bottom: Structure of KMnF3 which tends to form during heating of K2MnF6. | |
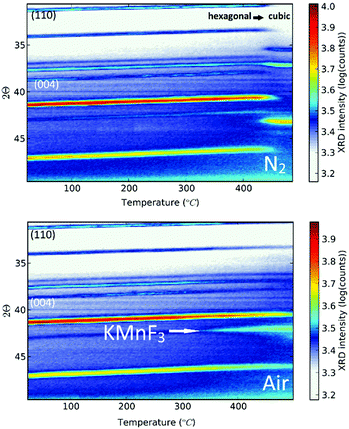 |
| | Fig. 2
In situ XRD spectrum of K2MnF6, heated from RT to 500 °C under a N2 atmosphere (top) or in air (bottom). A phase transformation is visible around 440 °C when N2 is used as an annealing atmosphere. No phase transformation is observed in air, but KMnF3 is slowly formed with increasing temperatures. | |
 |
| | Fig. 3
Ex situ XRD spectrum of K2MnF6 before heating (top) and after heating in air or N2 (middle and bottom, respectively). | |
Note that in Fig. 2, a large thermal shift towards considerable smaller values for 2θ is observed for all XRD peaks of the hexagonal K2MnF6 when the temperature is increased, indicating a rather large thermal expansion of the lattice. Since K2MnF6 is hexagonal below 400 °C, it can be expected that the positive thermal expansion of the lattice is anisotropic. The peak shifts can be related to the elongation of the lattice vectors a, b and c of the unit cell as the temperature is increased. Gaussian peak fitting of the (110) and (004) diffraction peaks at RT and 400 °C showed that the lattice has expanded in an anisotropic way with an estimated elongation of 2.1% along the c-axis, and only 1.6% along the a and b axes. Hence, anisotropic expansion might involve rotation of all [MnF6]2− octahedral complexes to ultimately align with the c-axis. This positive thermal expansion is next followed by negative thermal expansion in the vicinity of phase transition (see the N2 case in Fig. 2). Negative expansion could be caused by coupled rotation of rigid [MnF6]2− units when the temperature is close to the transition temperature. Perovskite structures are known to exhibit so-called rigid unit modes (RUMs).34,35 These are low-energy phonon modes in which the rigid units are rotationally coupled, often leading to contracting structures upon heating. Negative thermal expansion is an indicator for the existence of RUMs.
When, on the other hand, heating of K2MnF6 is performed in air instead of N2, a different behaviour is found. The bottom figure of Fig. 2 shows broadening of the peak at 2θ = 43° instead of phase transition. Remarkably, the intensity of this peak increases above 300 °C while the other peaks tend to fade out above this temperature. This is explained by the formation of KMnF3 (KF·MnF2) which also has a diffraction peak around 2θ = 43°. The structure of KMnF3 (lower part of Fig. 1) is similar to the cubic phase of K2MnF6, the difference being the doubled number of coordination complexes within a cubic unit cell, which are corner-sharing in the case of KMnF3 as a result of the reduction of Mn4+ to Mn2+. In this way, a perovskite structure appears. Although more pronounced in air, thermal degradation is still present to some degree when N2 is used, as can be verified in Fig. 3. As will be clarified later on, increased thermal degradation in air is due to the hygroscopic nature of K2MnF6. Water vapour present in air can lead to hydrolysis of the [MnF6]2− complexes which in turn facilitates the reduction of Mn4+ and hence the structural change. Probably, the formation of KMnF3 is accompanied by the evaporation of HF as the fluorine content is lowered in KMnF3 compared to K2MnF6. It is furthermore suspected that HF liberation can take place even at room temperature as a consequence of hydrolysis. This is evidenced by storing K2MnF6 in a glass vial under air. The vial gets eroded over time while the powder at the glass interface turns dark brown in a period of days up to weeks, probably depending on the purity of the K2MnF6 powder. We will elaborate on synthesis impurities in the next section. SEM-EDX measurements to investigate the brown discoloration showed the presence of Si and O as additional elements, indicating that they result from etching reactions at the glass interface. Nevertheless, the formed products could not be identified as a separate crystalline phase as the XRD pattern did not show any additional peaks next to K2MnF6.
Synthesis impurities
The excess KF that is used during synthesis ensures sufficient fluorination of Mn ions. Fluorination prevents excessive reduction towards Mn valences lower than 4+, which would be accompanied by the formation of oxygen containing or hydrated structures by interaction of Mn with H2O and H2O2 in the synthesis solution as will be discussed later on. As can be seen in Fig. 4, the end result after synthesis can contain several impurities after precipitation and drying. The main impurity proves to be KHF2, a corrosive salt that is often used as a glass etchant. The salt can result from the interaction of HF with KF in the synthesis solution:
| HF + KF → KHF2 (synthesis solution) |
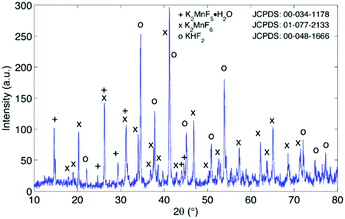 |
| | Fig. 4
Ex situ XRD pattern after K2MnF6 synthesis without washing treatment. KHF2 and K2MnF5·H2O formed during K2MnF6 synthesis as a result of KF–HF interaction and Mn–H2O or Mn–H2O2 interaction, respectively. | |
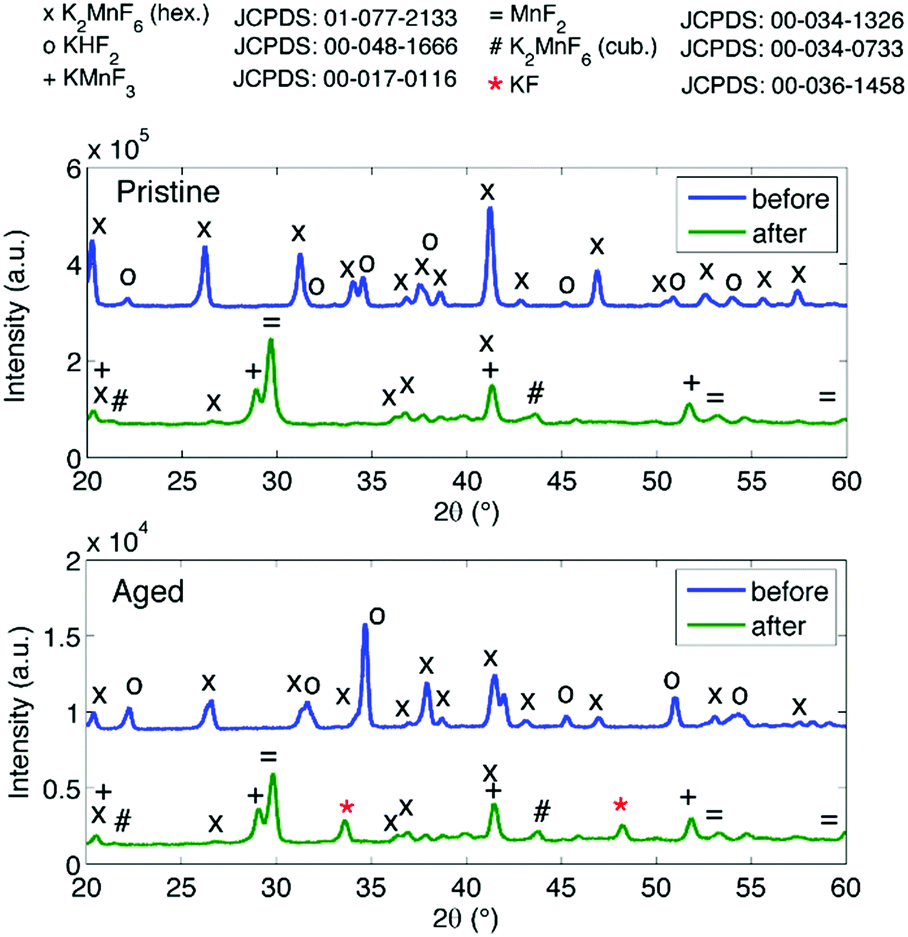
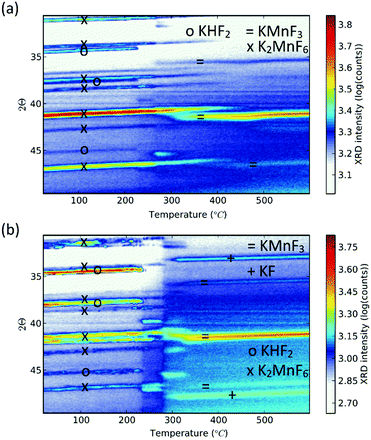
Purification requires an additional washing treatment with HF before drying, in this way removing any residual KHF2. The importance of proper purification of K2MnF6 should not be underestimated. Not only is the KHF2 impurity a hazardous corrosive compound on its own, but elevated temperatures also lead to the decomposition and melting of the phase which is accompanied by the evaporation of HF. The corrosive behaviour of this impurity could explain the observed corrosion of K2MnF6 powder stored in glass vials as was mentioned in the previous section. To investigate the effect of hydrolysis on samples containing K2MnF6 and KHF2, two samples were heated in air from RT up to 600 °C, one of which was aged in a weathering chamber first (see Fig. 5).
 |
| | Fig. 5
In situ XRD measurements on impure powders which were (a) fresh and (b) aged. Both samples contain K2MnF6 and KHF2 at room temperature as no washing step was performed during synthesis. KHF2 melts at 239 °C, after which decomposition causes HF evaporation. At high temperatures KMnF3 is formed. KF is additionally formed in (b) because the pristine amount of KHF2 was higher (see Fig. 6) and hydrolysis of KHF2 occurred during the ageing process. | |
The weathering conditions of the aged sample were 24 h at 85 °C/85% relative humidity (RH) followed by 48 h at 40 °C/40% RH. The XRD patterns of both samples before heat treatment can be found in Fig. 6, showing that the amount of KHF2 impurity before heating was larger in the aged sample. Despite the resulting intensity differences in the in situ XRD patterns of Fig. 5, the global thermal behaviour of both samples can be considered as equal. When the powders are heated above 239 °C, the powders start to release HF because KHF2 melts and decomposes into KF and HF at this temperature. Two relatively weak subsequent phase transitions occur after melting until crystalline KMnF3/KF·MnF2, KF and MnF2 are ultimately formed above 300 °C as can be verified in Fig. 5. A comparison of Fig. 5 with Fig. 2 indicates that even small amounts of KHF2 can drastically lower the decomposition temperature of K2MnF6. Note that two additional XRD peaks are present in the aged sample which are identified as KF resulting from the larger amount of KHF2 melt in the aged sample and hydrolysis of KHF2 during aging. In Fig. 4, one can observe a second synthesis-related impurity next to KHF2. As was shown by Kasa et al.,36 uncontrolled reduction of Mn can lead to the formation of a pink precipitate. In some of the powders synthesized in this work, pink coloured particles were occasionally formed next to the yellow K2MnF6 particles. The XRD analysis in Fig. 4 identified the pink phase as K2MnF5·H2O, in agreement with the work of Kasa et al.36 The structure of K2MnF5·H2O is shown in Fig. 7. Water molecules are incorporated in this crystalline structure; however the complexes are not attacked by the hydration process. Nevertheless, hydrate formation can impede further phosphor synthesis as Mn has been reduced to the 3+ valence state rather than the 4+ valence state.
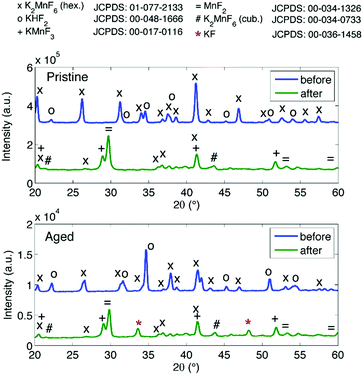 |
| | Fig. 6
Ex situ XRD measurements at RT on a pristine and aged impure powder before and after heating under an inert atmosphere. The same behaviour is present in both samples. Two additional peaks are seen in the aged sample, indicated by the red asterisks. These are due to KF formation because of the high pristine amount and hydrolysis of KHF2 in this sample. | |
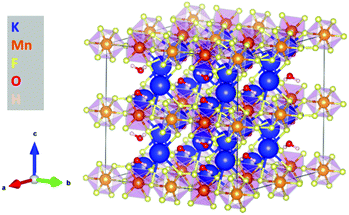 |
| | Fig. 7 Structure of the K2MnF5·H2O hydrate formed during synthesis. | |
Further phosphor synthesis: degradation in HF solution
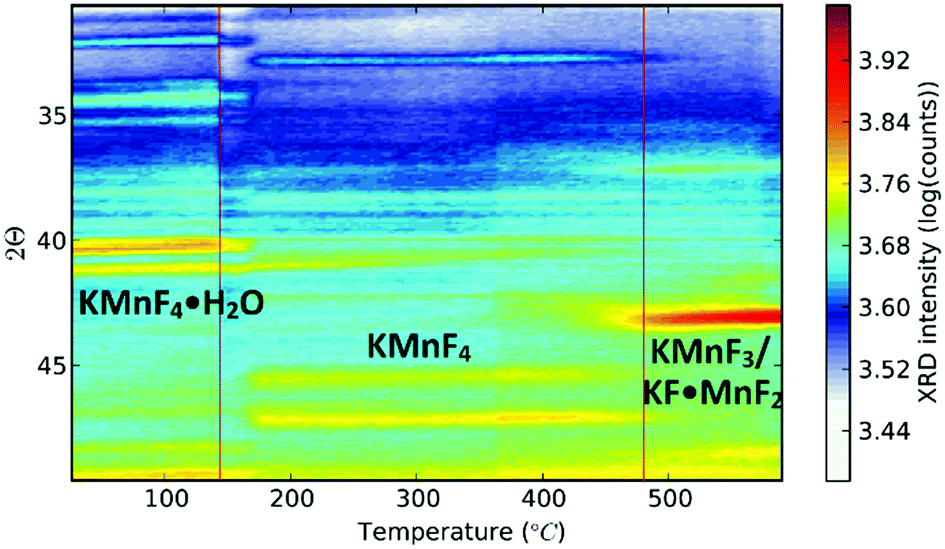
Up to this point, we discussed the risk of impurity phase formation as a result of KF–HF interaction, hydrolysis and hydration during synthesis. Moreover we discussed crystallization of KMnF3 when K2MnF6 is heated in air. Equally important is the stability of Mn4+ when the purified K2MnF6 is used as a precursor in further synthesis steps of fluoride phosphors. Typically the precursor material will be dissolved in a KF/HF solution which is brought to elevated temperatures even up to 60 °C.30 K2MnF6 dissociates into K+ and [MnF6]2−, making the latter vulnerable to hydrolysis. To study these effects, a purified K2MnF6 sample was dissolved into a 40% HF solution which was subsequently heated in a water bath at set temperatures. A colour change of the solution could be observed: the pristine yellowish solution at 20 °C turned dark brown at 55 °C, with a gradual change in between. The colour change indicates that the [MnF6]2− complexes were (at least partially) decomposed by the solution. To further investigate the effect of the solution temperature on the [MnF6]2− complexes, acetone was added as a nonsolvent to form precipitates at different temperatures. Analysis with ex situ XRD on the dried precipitates was performed to identify the dominant species formed at each temperature. The resulting XRD spectra of the precipitates are shown in Fig. 8. Phase identification indicates that the K2MnF6 powder gradually transforms into the KMnF4·H2O monohydrate structure when the K2MnF6 powder is dissolved in a heated 40% HF solution, with a greater conversion at higher temperatures. A comparison of the crystallographic structure of K2MnF6 with the structure of KMnF4·H2O, shown at the bottom of Fig. 8, reveals that half of the Mn complexes have been attacked by water molecules in the hydrate. These complexes lost two fluorine ligands, which are now replaced by water molecules. One ultimately ends up with one incorporated water molecule per Mn atom. The formation of these metal aqua-complexes in K2MnF6 is in accordance with ref. 37, in which hydrolysis reactions were considered to cause parasitic effects on the K2MnF6 luminescence. Hydrolysis and hydration could replace fluorine ions with water molecules, potentially forming oxygen octahedrons during K2MnF6 synthesis. These MnO6 octahedrons could explain the K2MnF6 luminescence above 680 nm observed at 75 K. As shown in Fig. 9, this emission is similar to the one found in Mn(IV) doped oxides where Mn4+ is coordinated by an oxygen octahedron. For Mn4+ in an octahedral fluorine environment, like in e.g. K2SiF6:Mn4+, the emission is blue-shifted by about 50 nm.15 Hydrolysis reactions are thus a major challenge when synthesizing and using K2MnF6 during fluoride phosphor synthesis. As with the H2O2 addition rate in K2MnF6 synthesis, the addition rate of the KF/K2MnF6/HF solution during the fluoride phosphor synthesis should also be fine-tuned to minimize these hydrolysis reactions that irreversibly reduce Mn4+ to Mn3+ and lead to hydrated structures. To verify whether the crystal water in KMnF4·H2O could be evaporated, KMnF4·H2O was heated in N2 up to 600 °C and followed using in situ XRD (Fig. 10). As can be seen, if the temperature is increased above 145 °C, the crystal water is evaporated from the hydrate, leaving KMnF4 behind. KMnF4 decomposes upon further heating into KMnF3 with the loss of fluorine, probably through HF liberation. As will be cleared out in the following section, the reduction of Mn4+ can cause parasitic optical absorption and might lower the efficiency of the final fluoride phosphor synthesized using the impure or degraded K2MnF6 precursor.
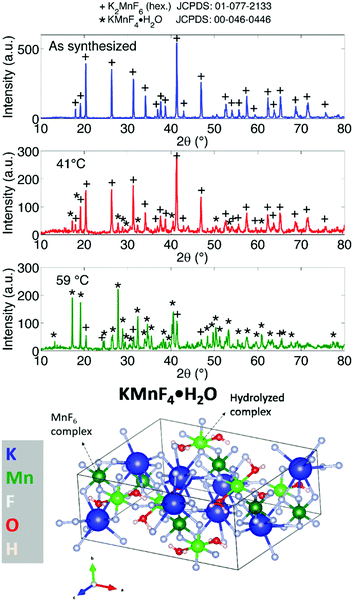 |
| | Fig. 8 (top) XRD pattern of dried precipitates produced by heating K2MnF6 in 40% HF solutions to set temperatures and the addition of acetone as a nonsolvent. (bottom) Structure of KMnF4·H2O. Some MnF6 complexes have two fluorine atoms substituted by H2O molecules. Dark green atoms are manganese atoms in MnF6 complexes. The light green Mn atoms are the centres of the hydrolysed complexes. | |
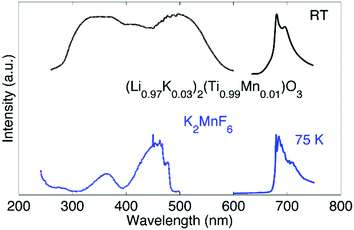 |
| | Fig. 9 Luminescence emission (solid line) and excitation (dotted line) spectra of (Li0.97K0.03)2(Ti0.99Mn0.01)O3 at RT (top, taken from ref. 38) and K2MnF6 at 75 K (bottom). | |
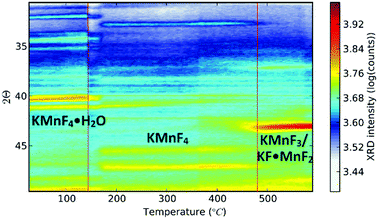 |
| | Fig. 10
In situ XRD measurement performed on the KMnF4·H2O monohydrate structure. As the temperature is increased water evaporates from the hydrate, leaving dehydrated KMnF4. Further heating produces the KMnF3 crystalline phase. | |
Diffuse reflectance and X-ray absorption of K2MnF6
The influence of Mn3+ formation on the optical absorption behaviour of K2MnF6 was investigated by diffuse reflectance measurements on MnF2, MnF3, (KMnF4·H2O/K2MnF6), K2MnF6, (K2MnF6/K2MnF5·H2O/KHF2) and aged K2MnF6. The parentheses indicate that the powder was a combination of multiple crystalline phases as was verified by XRD. For the aged sample, conditions of 60 °C and 50% RH were used. The complete diffuse reflectance spectra of the compounds are shown in Fig. 11a. Fig. 11b shows a more detailed part of the reflection patterns in the areas of interest. In MnF2, used as a Mn2+ reference here, a smooth broad absorption band is seen which increases on going from larger to smaller wavelengths. No sharp absorption features can be observed. On the other hand, clear signatures of Mn3+ are found in MnF3 (used as a Mn3+ reference). Two sharp minima are visible at 432 nm and 518 nm together with a broader absorption band around 800 nm, indicated by the arrow in Fig. 11a.
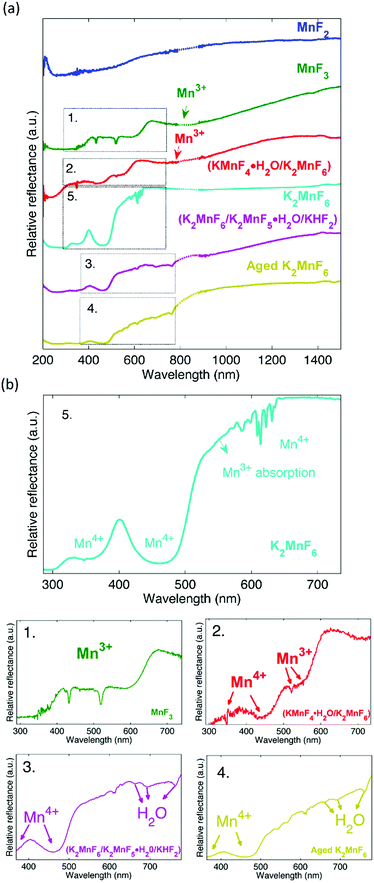 |
| | Fig. 11 (a) Diffuse reflectance measurement on diverse fluorides. The interpolated data in the region where the detector is switched are indicated by a dashed line. The spectra are displaced vertically for clarity. (b) Enlarged areas of the diffuse reflectance spectra in figure (a). | |
Furthermore, a drop in the diffuse reflectance is observed below 664 nm and below 413 nm, forming a staircase like feature in the reflectance spectrum. As expected, a similar behaviour is seen in (KMnF4·H2O/K2MnF6). This confirms the presence of Mn3+ in the hydrated compound. A comparison of the diffuse reflectance spectrum of (KMnF4·H2O/K2MnF6) with the diffuse reflectance of pure K2MnF6 reveals that Mn4+ absorption bands around 350 nm and 450 nm are also present. These absorption bands can be assigned to the 4A2g → 4T1g and 4A2g → 4T2g electronic transitions in Mn4+, respectively.39 In the case of (K2MnF6/K2MnF5·H2O/KHF2), the part of the broad Mn3+ absorption band above 800 nm can still be observed. However, below 800 nm the diffuse reflectance shows a different behaviour than was the case in the previously described Mn3+ containing compounds. If compared with the diffuse reflectance measurement on the aged K2MnF6, a similar behaviour is observed. Likely, absorbed water in the aged K2MnF6 and K2MnF5·H2O influences the optical absorption behaviour below 800 nm of both compounds. To conclude, indications exist that even purified K2MnF6, without traces of parasitic crystalline phases in XRD, can still contain a considerable fraction of Mn3+ ions. The 450 nm absorption band corresponding to Mn4+ in K2MnF6 is accompanied by an extra absorption band on the longer wavelength side, which is superposed on the sharp Mn4+ absorption lines around 620 nm. A comparison with the MnF3 and (KMnF4·H2O/K2MnF6) spectra suggests that this broad absorption might be due to a fraction of the Mn ions that are in the 3+ valence state. To verify this, XANES measurements on Mn foil, MnF2, MnF3 and K2MnF6 were performed. The XANES results are shown at the top of Fig. 12. Here, it is clear that the absorption edge energies shift towards higher energy values as the oxidation state of Mn is increased from 0 (Mn foil) towards 4+ (K2MnF6). To determine the edge position of each compound more accurately, the first derivative spectra, shown in the middle of Fig. 12, are used. In the case of MnF2, MnF3 and K2MnF6, the global maximum is taken as the K-edge position for the octahedral fluorine coordination of Mn2+, Mn3+ and Mn4+, respectively. However in the case of pure Mn foil the first maximum is taken as the edge position for neutral Mn. The reason behind this will be explained later on. If the absorption edge energy is plotted against the assumed dominant Mn valence in MnF2, MnF3 and K2MnF6, all having similar octahedral fluorine coordination of Mn, a linear relationship is obtained. This confirms the dominant contribution of Mn2+, Mn3+ and Mn4+ in MnF2, MnF3 and K2MnF6, respectively. However, as Mn is vulnerable to valence changes, multivalence of the Mn ions in the fluoride powders is probable. Partial reduction and oxidation of the Mn within the fluoride powders indeed contributes to the XANES spectra. As can be seen in the MnF2 derivative curve, small contributions of neutral and trivalent Mn can be found as relative maxima at the edge positions of neutral and trivalent Mn. Likewise, the MnF3 first derivative curve shows relative maxima at the edge energies of neutral and tetravalent manganese, again indicating partial reduction and oxidation of the Mn ions in MnF3. In the Mn foil, we ascribe the additional peaks at higher energies in the first derivative spectrum to oxidized Mn. Probably contact with air caused the (surface) formation of diverse manganese oxides. In K2MnF6, a second smaller maximum is found in the derivative spectrum which moreover coincides with the global Mn3+ maximum. Hence it is likely that a fraction of the Mn ions in K2MnF6 is in a 3+ valence state rather than the desired 4+. Since no traces were found in XRD, this indicates that the reduction of Mn4+ to Mn3+ does not always involve crystalline phase formation.
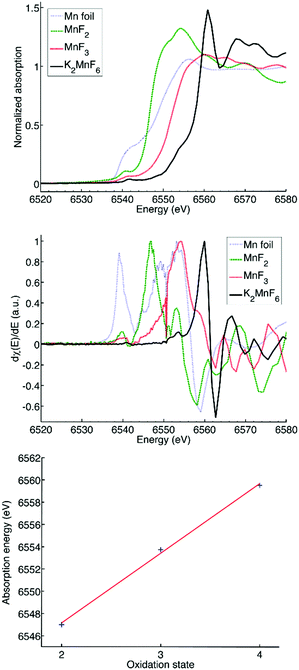 |
| | Fig. 12 Top: XANES measurement on Mn foil, MnF2, MnF3 and K2MnF6. Middle: First derivative spectra of the curves in the top figure. Bottom: Linear fit of the absorption edge energy as a function of the Mn oxidation state. | |
Based on a Gaussian peak fit of the Mn3+ and Mn4+ derivative XANES spectra, the fraction of trivalent manganese ions in K2MnF6 can be estimated to be smaller than 20%. Remarkably, no clear indications of neutral Mn and Mn2+ were found, confirming that the initial synthesis related hydrolysis processes predominantly lead to the formation of Mn3+. Mn2+ on the other hand is formed by hydrolysis in air at higher temperatures as was shown in the section concerning the structural properties of K2MnF6.
Conclusions
K2MnF6 is an essential precursor in the two-step co-precipitation synthesis of Mn4+ doped red-emitting fluoride phosphors. K2MnF6 incorporates Mn4+ in a fluorine octahedron, thereby stabilizing the 4+ valence state of Mn, as required. Nevertheless, the purity of K2MnF6 is an issue as both synthesis and degradation by hydrolysis and heat produce several impurities. This can in turn cause defects and impurities in the final phosphor, thereby worsening thermal quenching and lowering the phosphor efficiency. Firstly, the synthesis of K2MnF6 is impeded by the formation of KHF2, a highly corrosive salt which melts at 239 °C. It is caused by the interaction of KF with HF in the synthesis solution. KHF2 should be washed out by an additional washing step in HF before drying the precipitate. A second synthesis related problem arises with insufficient control of the Mn reduction in the synthesis solution. Any excess of H2O2 can lead to excessive reduction of Mn, often leading to hydrated structures such as K2MnF5·H2O. Furthermore, diffuse reflectance, XAS and XRD showed that the presence of Mn3+ does not a priori lead to crystalline impurity phases such as a hydrate structure. Even when a pure K2MnF6 powder is obtained, further synthesis steps may cause a valence change of the Mn4+ ions. Hydration and hydrolysis can take place when K2MnF6 is dissolved in heated HF solutions which are used in phosphor synthesis. KMnF4·H2O was found in XRD measurements as an impurity after dissolving K2MnF6 in 40% HF at elevated temperatures. The pristine [MnF6]2− complexes in K2MnF6 have been hydrolysed, resulting in water molecules which take the place of fluorine. In this way, KMnF4·H2O is formed. Moreover, Mn4+ reduces to Mn3+, the latter affecting the optical properties of the final phosphor materials. Apart from synthesis difficulties, care should also be taken in the storage of K2MnF6. The hygroscopic nature of K2MnF6 shows that this compound is easily hydrolysed by water vapour in air, as could be seen by the large difference when air or N2 is used as an annealing atmosphere. Likely, hydrolysis facilitates Mn4+ to Mn2+ reduction when the temperature is increased. The final 2+ valence state was detected in XRD as KMnF3/KF·MnF2 is formed at temperatures above 300 °C. Therefore, we consider hydrolysis reactions as the main factor impeding the structural stability of K2MnF6. The hygroscopic nature of K2MnF6 in air and the tendency to form hydrated structures in solutions can severely influence the powder purity during use in the lab.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
RV, HFS, KK, DP and PFS thank the agency for Innovation by Science and Technology (IWT) for a SBO-IWT grant (SBO130030). Our gratitude also goes out to the staff and infrastructure at the DUBBLE beamline, ESRF, Grenoble, France (proposal 26-01-1110).
References
- S. Ye, F. Xiao, Y. X. Pan, Y. Y. Ma and Q. Y. Zhang, Mater. Sci. Eng., R, 2010, 71, 1–34 CrossRef.
- N. Narendran, Y. Gu, J. P. Freyssinier, H. Yu and L. Deng, J. Cryst. Grow., 2004, 268, 449–456 CrossRef CAS.
- G. Q. Zhang, W. Dirk van Driel and M. Yazdan Mehr, Engineering, 2015, 1, 170–178 CrossRef.
- P. F. Smet, A. B. Parmentier and D. Poelman, J. Electrochem. Soc., 2011, 158, R37–R54 CrossRef CAS.
- A. Piquette, W. Bergbauer, B. Galler and K. C. Mishra, ECS J. Solid State Sci. Technol., 2015, 5, R3146–R3159 CrossRef.
- Q. Zhou, H. Y. Tan, Y. Y. Zhou, Q. H. Zhang, Z. L. Wang, J. Yan and M. M. Wu, J. Mater. Chem. C, 2016, 4, 7443–7448 RSC.
- International Telecommunication Union, 2012.
- J. H. Oh, Y. J. Eo, H. C. Yoon, Y. D. Huh and Y. R. Do, J. Mater. Chem. C, 2016, 4, 8326–8348 RSC.
- Y. H. Kim, P. Arunkumar, B. Y. Kim, S. Unithrattil, E. Kim, S.-H. Moon, J. Y. Hyun, K. H. Kim, D. Lee, J.-S. Lee and W. B. Im, Nat. Mater., 2017, 16, 543–550 CrossRef CAS PubMed.
- Y. Hu, W. Zhuang, H. Ye, S. Zhang, Y. Fang and X. Huang, J. Lumin., 2005, 111, 139–145 CrossRef CAS.
- X. Piao, K. Machida, T. Horikawa, H. Hanzawa, Y. Shimomura and N. Kijima, Chem. Mater., 2007, 19, 4592–4599 CrossRef CAS.
- Y. Q. Li, J. E. J. van Steen, J. W. H. van Krevel, G. Botty, A. C. A. Delsing, F. J. DiSalvo, G. de With and H. T. Hintzen, J. Alloys Compd., 2006, 417, 273–279 CrossRef CAS.
- Z. Zhou, N. Zhou, M. Xia, M. Yokoyama and H. T. Hintzen, J. Mater. Chem. C, 2016, 4, 9143–9161 RSC.
- H. F. Sijbom, J. J. Joos, L. I. D. J. Martin, K. Van den Eeckhout, D. Poelman and P. F. Smet, ECS J. Solid State Sci. Technol., 2016, 5, R3040–R3048 CrossRef CAS.
- H. F. Sijbom, R. Verstraete, J. J. Joos, D. Poelman and P. F. Smet, Opt. Mater. Express, 2017, 7, 3332–3365 CrossRef.
- X. Y. Jiang, Y. X. Pan, S. M. Huang, X. Chen, J. G. Wang and G. K. Liu, J. Mater. Chem. C, 2014, 2, 2301–2306 RSC.
- S. Adachi, H. Abe, R. Kasa and T. Arai, J. Electrochem. Soc., 2012, 159, J34–J37 CrossRef CAS.
- M. Du, F. Tang, J. Long, C. Ma, X. Yuan, J. Zhang, Z. Wen, R. Ma and Y. Cao, Mater. Res. Bull., 2016, 83, 316–323 CrossRef CAS.
- H. Zhu, C. C. Lin, W. Luo, S. Shu, Z. Liu, Y. Liu, J. Kong, E. Ma, Y. Cao, R. S. Liu and X. Chen, Nat. Commun., 2014, 5, 4312 CAS.
- Q. Zhou, Y. Y. Zhou, Y. Liu, Z. L. Wang, G. Chen, J. H. Peng, J. Yan and M. M. Wu, J. Mater. Chem. C, 2015, 3, 9615–9619 RSC.
- S. Adachi and T. Takahashi, J. Appl. Phys., 2009, 106, 013516 CrossRef.
- Y. K. Xu and S. Adachi, J. Appl. Phys., 2009, 105, 013525 CrossRef.
- Q. Zhou, Y. Y. Zhou, Y. Liu, L. J. Luo, Z. L. Wang, J. H. Peng, J. Yan and M. M. Wu, J. Mater. Chem. C, 2015, 3, 3055–3059 RSC.
- S. Adachi and T. Takahashi, J. Appl. Phys., 2008, 104, 023512 CrossRef.
- L. Lv, X. Jiang, S. Huang, X. A. Chen and Y. Pan, J. Mater. Chem. C, 2014, 2, 3879 RSC.
- B.-E. Yeo, Y.-S. Cho and Y.-D. Huh, Opt. Mater., 2016, 51, 50–55 CrossRef CAS.
- D. H. Kim, J. Ju and D. Y. Jeong, New Physics: Sae Mulli, 2016, 66, 311–316 CrossRef.
- A. A. Setlur, E. V. Radkov, C. S. Henderson, J.-H. Her, A. M. Srivastava, N. Karkada, M. S. Kishore, N. P. Kumar, D. Aesram, A. Deshpande, B. Kolodin, L. S. Grigorov and U. Happek, Chem. Mater., 2010, 22, 4076–4082 CrossRef CAS.
- T. T. Deng, E. H. Song, J. Sun, L. Y. Wang, Y. Deng, S. Ye, J. Wang and Q. Y. Zhang, J. Mater. Chem. C, 2017, 5, 2910–2918 RSC.
- L.-L. Wei, C. C. Lin, M.-H. Fang, M. G. Brik, S.-F. Hu, H. Jiao and R.-S. Liu, J. Mater. Chem. C, 2015, 3, 1655–1660 RSC.
- F. Tang, Z. Su, H. Ye, M. Wang, X. Lan, D. L. Phillips, Y. Cao and S. Xu, J. Mater. Chem. C, 2016, 4, 9561–9568 RSC.
- H. J. H. Bode and F. Bandte, Angew. Chem., 1953, 304 CrossRef CAS.
- P. F. Smet and J. J. Joos, Nat. Mater., 2017, 16, 500–501 CrossRef CAS PubMed.
- A. P. Giddy, M. T. Dove, G. S. Pawley and V. Heine, Acta Crystallogr., Sect. A: Found. Crystallogr., 1993, 49, 697–703 CrossRef.
- M. T. Dove, Am. Mineral., 1997, 82, 213–244 CAS.
- R. Kasa, Y. Arai, T. Takahashi and S. Adachi, J. Appl. Phys., 2010, 108, 113503 CrossRef.
- A. M. Black and C. D. Flint, J. Chem. Soc., Dalton Trans., 1974, 15, 977–981 RSC.
- S. K. K. Seki, K. Uematsu, T. Ishigaki, K. Toda and M. Sato, Journal of Ceramic Processing Research, 2013, 14, s67–s70 Search PubMed.
- R. Kasa and S. Adachi, J. Electrochem. Soc., 2012, 159, J89–J95 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Heleen F.
Sijbom
ab,
Heleen F.
Sijbom