
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Processing of ferroelectric polymers for microelectronics: from morphological analysis to functional devices†
Hamed Sharifi
Dehsari
,
Jasper J.
Michels
 * and
Kamal
Asadi
*
* and
Kamal
Asadi
*
Max Planck Institute for Polymer Research, Ackermannweg 10, 55128, Mainz, Germany. E-mail: michels@mpip-mainz.mpg.de; asadi@mpip-mainz.mpg.de
First published on 14th August 2017
Abstract
Solution casting under ambient conditions of thin films of the ferroelectric copolymer poly(vinylidene fluoride-co-trifluoroethylene) (P(VDF-TrFE)) is highly attractive for cost-effective production of flexible memory devices. However, rough and porous films obtained under ambient conditions typically give a low yield of working devices. A major challenge is ambient water vapor condensing into the drying solution, causing non-solvent vapor-induced phase separation (VIPS). By integrating solution-stage modeling, microscopic analysis and thin-film device characterization, we show that the hydrophilicity of solvent is a deciding factor in obtaining properly functioning capacitive memory elements based on P(VDF-TrFE) under ambient conditions. Our numerical study, involving the ternary phase diagram of the polymer/water/solvent blend as well multicomponent dynamic phase field modeling, predicts the occurrence of VIPS for a given solvent hygroscopicity and demonstrates an increase in early stage domain size with decreasing relative humidity, whereas the opposite trend is predicted for the rate of demixing. Experimentally observed morphologies are consistent with the numerical simulations. For a sufficiently low solvent hygroscopicity, >90% production yield of devices operating at voltages on par with upscaled thin-film flexible electronics is achieved.
Introduction
Non-volatile memory based on solution-cast thin films of ferroelectric polymers has emerged as a promising low-cost information storage technology for large-area microelectronic applications.1,2 The random copolymer poly(vinylidene-fluoride-co-triflouroethylene) (P(VDF-TrFE)) is the most attractive material due to its high remanent polarization3–5 and therefore frequently used in memory elements, such as capacitors,6,7 ferroelectric field-effect transistors8,9 and diodes.10–13 To write/erase data, ferroelectric polarization is switched by an external electric field that is larger than the coercive field, Ec, which for P(VDF-TrFE) amounts to ∼50 MV m−1.4,5 In principle, this high coercive field of P(VDF-TrFE) requires high voltages for polarization switching. To nevertheless keep the operation voltage below 10 V, polymer films with a thickness below 200 nm are required. However, since thin layer-devices are more susceptible to film imperfections, such as a rough microstructure, production yields are prohibitively suppressed.Limited device yield and concomitant large scatter in coercive voltage hinder upscaling of integrated arrays of P(VDF-TrFE) memory elements, especially under ambient conditions (20 °C, 50% relative humidity (RH)). Generally, P(VDF-TrFE) is cast from dimethylformamide (DMF) or ketones, which are often hygroscopic. For the homopolymer poly(vinylidene fluoride) (PVDF), it has been shown that condensation of ambient water into the DMF solution causes vapor-induced phase separation (VIPS), as water is a non-solvent for PVDF.14,15 The resulting domain structure prohibitively compromises the integrity and smoothness of PVDF films. Smooth PVDF films have so far only been obtained under an inert atmosphere,15–18 or at an elevated substrate temperature,15,19 conditions that are not beneficial for cost-effective production. VIPS is also expected to occur for P(VDF-TrFE) since its properties are similar to PVDF. Although avoiding processing-related film defects is still a major challenge for sub-200 nm thin-films, consistent studies considering the full spectrum of material properties, processing conditions, microstructure formation and thin film device performance are unfortunately still lacking.
The present work aims to lift this hiatus and provides novel insight into the dynamics of the early stages of VIPS, and how these relate to dry film integrity and device performance. This work covers all aspects of device preparation, starting with modeling and simulation of VIPS to pre-estimate solution behavior, proceeds with detailed microstructural analysis of cast P(VDF-TrFE) films and finally discusses device fabrication and electrical characterization. As such, a direct link is established between material properties, processing conditions and device performance. The generality of our approach makes the work not only relevant to the field of thin-film electronics, but hopefully to any technology relying on polymer/solvent/non-solvent processing, perhaps most prominently the manufacture of microporous membranes.
We consider P(VDF-TrFE) thin film processing from three different solvents with different hygroscopicities: DMF, cyclohexanone and cyclopentanone. Calculation of the ternary phase diagrams, combined with numerical simulation of structure formation under simultaneous solvent evaporation and water vapor dissolution, not only predicts the occurrence of VIPS as a function of solvent polarity, but also shows how initial domain size and demixing dynamics depend on ambient humidity. The calculations are validated by subjecting wire-bar coated P(VDF-TrFE) films under conditions of controlled temperature and relative humidity to extensive atomic force microscopy (AFM) analysis. Using the insight gained, we fabricate ferroelectric capacitors under ambient conditions, i.e. 20 °C, 50% RH, to establish how device yield and the scattering in coercive voltage are influenced by solvent polarity and ambient humidity.
Experimental
Materials
Ferroelectric copolymer P(VDF-TrFE) (65–35% molar) with molecular weight Mn = 140![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (PDI = 2.5) was purchased from Solvay. Poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate) (PEDOT:PSS) was purchased from Clevios. N,N-Dimethylformamide (DMF), anhydrous DMF (a-DMF) (≥99.9%), cyclohexanone and cyclopentanone were purchased from Sigma Aldrich. All the chemicals were used as-received without further purification.
000 (PDI = 2.5) was purchased from Solvay. Poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate) (PEDOT:PSS) was purchased from Clevios. N,N-Dimethylformamide (DMF), anhydrous DMF (a-DMF) (≥99.9%), cyclohexanone and cyclopentanone were purchased from Sigma Aldrich. All the chemicals were used as-received without further purification.
Film preparation
P(VDF-TrFE) was dissolved in different solvents (DMF, a-DMF, cyclohexanone and cyclopentanone) at a concentration of 100 mg ml−1. Glass substrates were cleaned by scrubbing in soap-water and subsequently sonicated for 10 minutes in deionized water, acetone and propanol, respectively. After drying at 120 °C for 10 minutes, the substrates were treated for 5 minutes with UV-O3. P(VDF-TrFE) films were coated under humidity-controlled conditions from solution using a wire-bar coating (K202 control coater, RK Print). Substrate temperature was varied between 20 °C and 80 °C. The relative humidity was adjusted between 10% and 80%. Film thicknesses were measured using a Dektak profilometer. The polymer films for morphological analysis were cast using the same initial solution volume of 100 μL and the same wire bar diameter. Since the initial concentration is constant, each coated film hence contained the same amount of polymer material.15 Polymer films prepared for devices were all tuned to have the same apparent mean thickness. This was accomplished by adjusting the concentration of the casting solution.Sample preparation for specific AFM analysis
To probe the morphology at the polymer/substrate interface, a sacrificial layer of PEDOT:PSS (∼80 nm) was spin coated and dried in a vacuum at 100 °C for 10 minutes prior to P(VDF-TrFE). After spin coating of the P(VDF-TrFE) layer, the substrates were submerged in deionized water to dissolve the PEDOT:PSS. The P(VDF-TrFE) films were picked up-side-down using a piece of mica. Both polymer/air and polymer/substrate interfaces were probed using atomic force microscopy (AFM) (Nanoscope Dimension 3100 Bruker). The topography and roughness were characterized before and after annealing at 140 °C for an area of 30 μm × 30 μm.Thin-film devices
Ferroelectric capacitors were fabricated by thermal evaporation of Au (70 nm) on the substrates using the Cr (2 nm) adhesion interlayer. Contact lines were defined using shadow masks. P(VDF-TrFE) thin films were deposited under ambient conditions (20 °C and 50% RH). The nominal layer thickness was tuned to be 150 ± 20 nm for all the solvents via changing the concentration of the polymer solution. After annealing at 140 °C for 2 h in a vacuum, a 70 nm Au top electrode was evaporated through a shadow mask. Electric displacement versus electric field D–E loops were measured under ambient conditions using a Sawyer-Tower circuit.Results and discussion
Model predictions
In order to pre-estimate whether the water-affinity of the solvent could cause VIPS to occur during solution casting under ambient conditions, we calculated for three different solvent polarities the isothermal phase diagrams of the ternary blend of P(VDF-TrFE) polymer (“P”)/water (= “non-solvent”) (“W”)/solvent (“S”), as well as the composition trajectories drawn through the phase diagram upon simultaneous solvent evaporation and water vapor dissolution. As mentioned above, three solvents were considered: DMF, cyclohexanone, and cyclopentanone, all known to be good solvents for P(VDF-TrFE). In particular, cyclohexanone is often used for the fabrication of P(VDF-TrFE)-based capacitors and memory diodes.11,20 DMF is fully miscible with water,21 whereas cyclohexanone and cyclopentanone exhibit partial miscibility: 86 g and 9 g per liter water at 20 °C, respectively.22,23 The phase diagrams were calculated using Flory-Huggins theory24,25 based on input parameters, i.e. binary interaction parameters χij and relative molecular size N, either derived directly from literature, or matched so as to represent the above given water–solvent miscibilities (see the ESI† for details). The calculated diagrams, plotted in Fig. 1, are consistent with previously reported (experimental) ones for polymer/non-solvent/solvent blends.14,26,27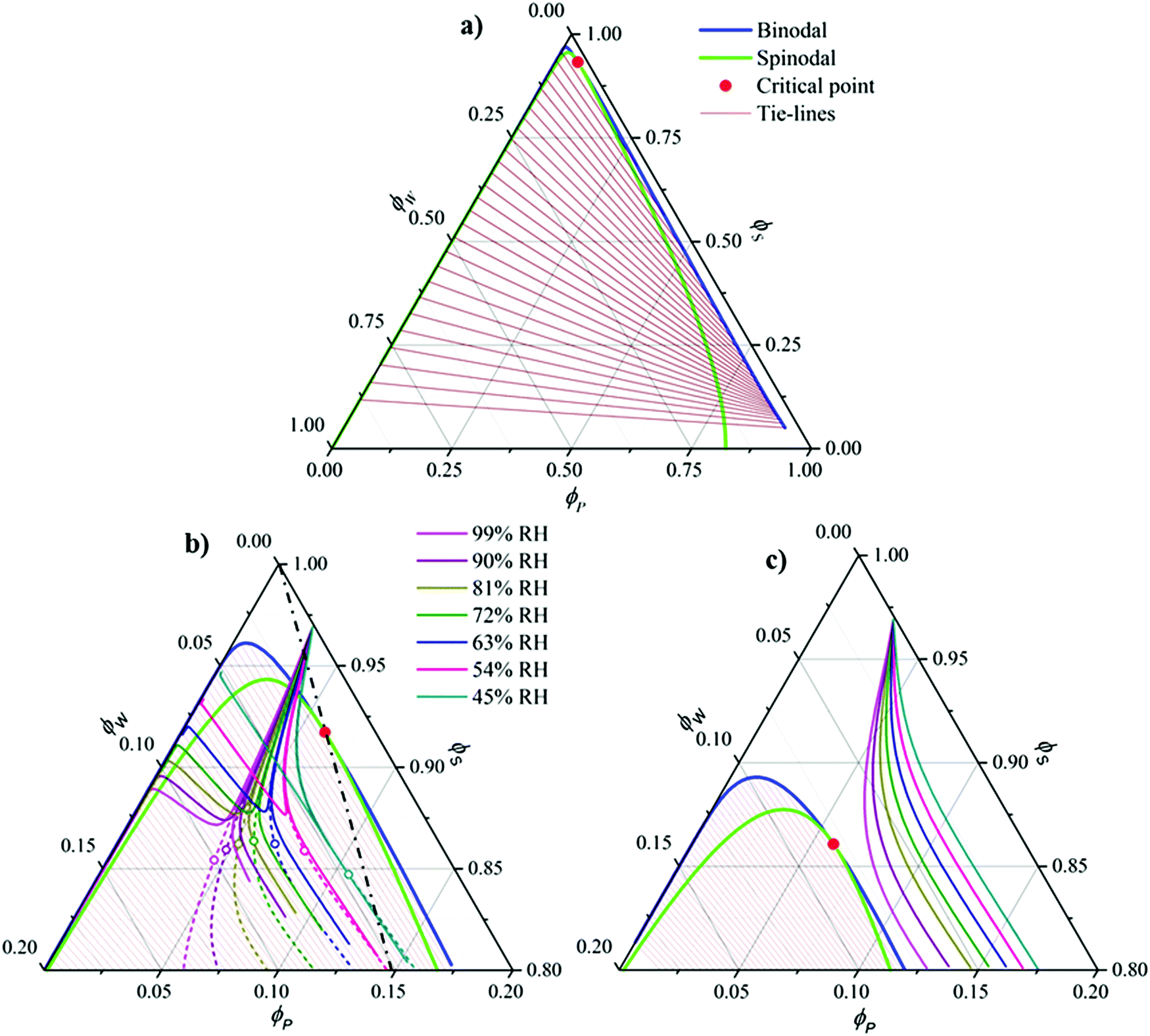 | ||
| Fig. 1 Calculated ternary phase diagrams for polymer (P)/water (W)/solvent (S) blends with S representing (a) DMF, (b) cyclohexanone, and (c) cyclopentanone. For (b) and (c) only the high-solvent region is shown. The curved colored lines in (a) and (b) indicate the numerically simulated composition trajectories associated with exposing an initially dry 3 vol% polymer solution to an atmosphere carrying a relative humidity in the range of 45–99%. In (b) demixing occurs, where the open symbols indicate the mean composition (dashed) at which the mixtures phase separate. The dash-dotted black line represents critical mean compositions, for which a bicontinuous structure is expected upon demixing. | ||
The phase diagram of DMF (Fig. 1a) is shown completely, whereas in the case of the phase diagrams of cyclohexanone and cyclopentanone (Fig. 1b and c), which are of comparable nature to that of DMF, only the top part is shown for the sake of clarity. The phase diagrams are characteristic of a polymer/non-solvent/solvent ternary, evidenced by (i) a large miscibility gap, (ii) a pronounced asymmetry, and (iii) steeply tilted tie-lines. The first feature is mainly due to the high value of the polymer–nonsolvent interaction parameter (χPW = 2.8, see ESI†). The asymmetry is a result of the difference in molecular size between a polymer and a water molecule and the tilt in the tie lines shows that if VIPS occurs, the solvent strongly prefers the polymer-poor phase. Fig. 1c shows that a lower solvent polarity somewhat reduces the miscibility gap.
The colored curves in Fig. 1b and c represent calculated (mean) composition trajectories resulting from simultaneous solvent evaporation and water ingress from/into the drying polymer solution, at various RH levels (see the legend). These curves were calculated assuming the volume flux σi of a blend component i across the liquid–vapor interface to be governed by the difference between its partial pressure just above the film and the applied partial pressure p∞i away from the vapor–liquid interface. Just above the drying film, a liquid–vapor equilibrium is assumed, so that the fluxes of water and solvent are given by:
 | (1) |
 the vapor pressure of the pure substance (tabulated for all solvents and water, see the ESI†), and k the mass transfer coefficient expressed in units of Pa−1 s−1 and for simplicity taken equal for water and solvent. The coefficient matrix on the RHS of eqn (1) enforces incompressibility. By defining
the vapor pressure of the pure substance (tabulated for all solvents and water, see the ESI†), and k the mass transfer coefficient expressed in units of Pa−1 s−1 and for simplicity taken equal for water and solvent. The coefficient matrix on the RHS of eqn (1) enforces incompressibility. By defining 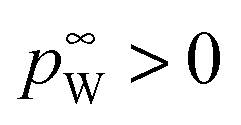 and
and 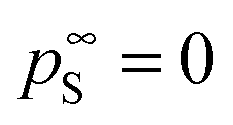 and by combining eqn (1) with the generalized 2D-diffusion model we published earlier,25,28,29 we not only track composition as a function of time, but also model the actual dynamics of structure formation due to VIPS.
and by combining eqn (1) with the generalized 2D-diffusion model we published earlier,25,28,29 we not only track composition as a function of time, but also model the actual dynamics of structure formation due to VIPS.
The calculations (Fig. 1b and c) assume an initially dry solution with a polymer concentration of 3 vol%. In the case of cyclohexanone the mean composition (dashed lines) readily enters the spinodal region, whereas for the more hydrophobic cyclopentanone, the trajectories remain within the single phase region. The calculations hence predict that for cyclohexanone spinodal VIPS occurs, but probably not for cyclohexanone. For DMF the results were similar to cyclohexanone and are hence not explicitly shown. We further note that although the model's simplifications (i.e. reduction by one space dimension and assuming a fixed mass transfer coefficient) together with the, at best, semi-quantitative nature of Flory–Huggins theory,30 does not allow for a fully quantitative description. However, trends regarding the dependence of demixing dynamics and initial domain size on relative humidity can certainly be identified and will be discussed below.
Fig. 1b (solid lines) shows that VIPS results in two coexisting liquid phases, one poor/rich and one rich/poor in polymer/solvent.31 Interestingly, the water fraction is similar in both phases due to the fact that the tie-lines run almost parallel to the solvent axis of the phase diagram. The shift in the bifurcation point (open symbols) with relative humidity suggests that the dynamics of VIPS, as well as the initial liquid-stage domain size, depends on the ambient conditions. This is shown in Fig. 2, which plots (Fig. 2a) composition ν as a function of dimensionless time ![[t with combining tilde]](https://www.rsc.org/images/entities/i_char_0074_0303.gif) (see the caption for definition), as well as the initial liquid-stage morphology for various relative humidities (Fig. 2b–e). While the demixing time scale increases with decreasing relative humidity, the opposite trend is observed for the initial domain size. This is explained by the fact that with decreasing humidity the bifurcation point approaches the spinodal and at the same time moves towards criticality (dash-dotted black line in Fig. 1b).
(see the caption for definition), as well as the initial liquid-stage morphology for various relative humidities (Fig. 2b–e). While the demixing time scale increases with decreasing relative humidity, the opposite trend is observed for the initial domain size. This is explained by the fact that with decreasing humidity the bifurcation point approaches the spinodal and at the same time moves towards criticality (dash-dotted black line in Fig. 1b).
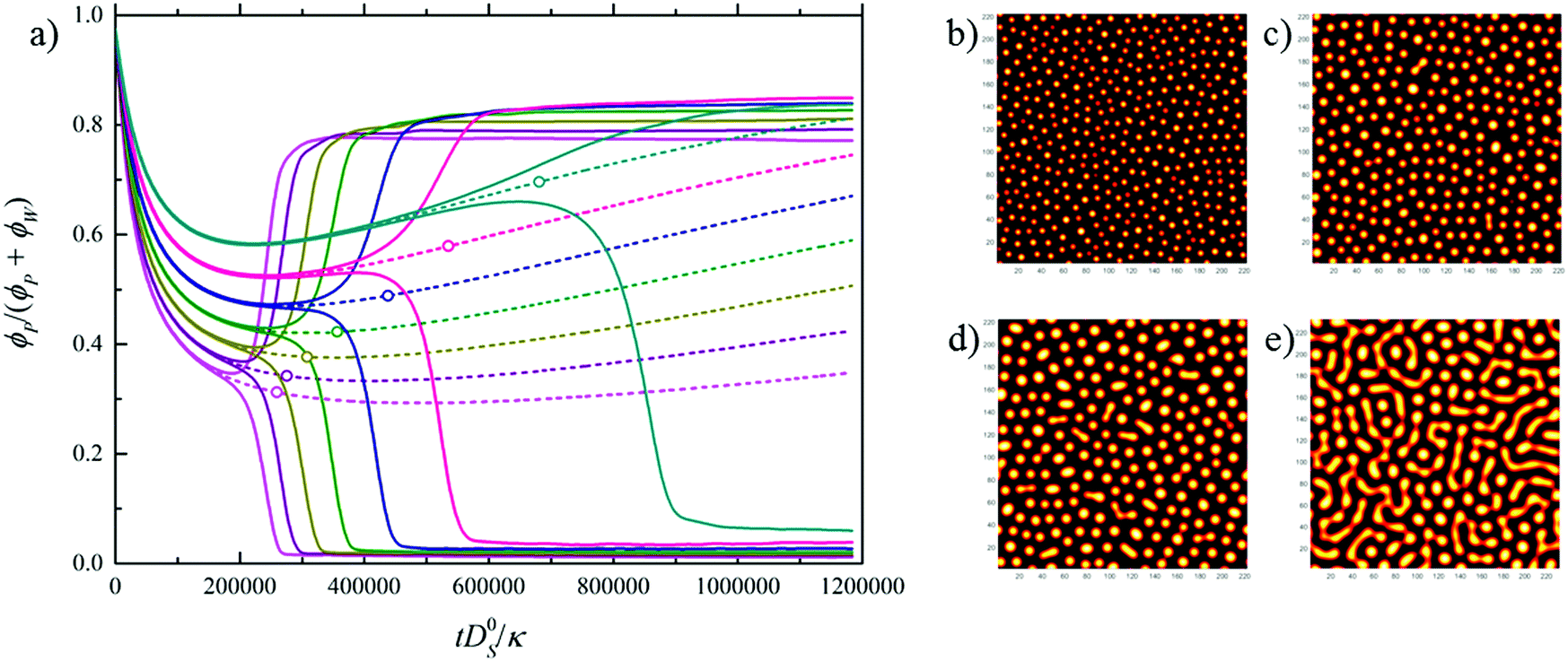 | ||
| Fig. 2 Numerical simulation of water vapor-induced spinodal decomposition of P(VDF-TrFE) in cyclohexanone for RH = 99–54%. (a) Composition ν = ϕP/(ϕP + ϕW) plotted as a function of dimensionless time = tD0S/κ, with D0S the self-diffusivity of the solvent (in m2 s−1) and κ (in m2) a thermal energy-normalized stiffness coefficient determining the liquid–liquid interface width30 (see the ESI†). (b–e) Initial liquid-stage demixed morphology (yellow = polymer rich, black = polymer devoid) corresponding to 99%, 72%, 63%, and 54% RH. | ||
In summary, going from high to low humidity a transition takes place from a structure wherein the polymer rich domains are dispersed in a solvent-rich matrix towards a more bicontinuous morphology of the polymer- and solvent-rich phases. At high relative humidity droplets are small and concentrated in the polymer, whereas at low humidity the polymer-rich phase consists of relatively large droplets in which the solid concentration is low. We emphasize that although models for nonsolvent-VIPS have been proposed,14,32 the level of detail provided by combining the calculated ternary phase diagram with numerical simulations of the demixing dynamics and associated structural evolution has so far not been given.
Microscopic analysis
As a next step we performed microscopic (AFM) analysis on dry P(VDF-TrFE) films cast from the above mentioned solvents under controlled humidity and substrate temperature conditions. Procedural details can be found in the Experimental section. Only representative data are reported below to substantiate the discussion. The ESI† contains a full account of all casting experiments and AFM analyses. Fig. 3 shows AFM topology images of films cast from wet (i.e. equilibrated) and anhydrous DMF under various conditions (see the caption). Fig. 3a, c and d show that at a substrate temperature of 20 °C the polymer layers consist of impinged micron-sized drop-like domains having considerable topological gradients, irrespective of the RH. For these films a root mean square (rms) roughness of ∼50 nm was obtained. In contrast, at a substrate temperature of 65 °C the films show much smaller features and are significantly smoother (Fig. 3b), with the rms roughness decreasing to ∼5 nm. | ||
| Fig. 3 Tapping mode AFM height images of P(VDF-TrFE) thin films coated at (a) DMF, 20 °C and 50% relative humidity, (b) 65 °C and 50% relative humidity, (c) DMF, 20 °C and 10% relative humidity, and (d) a-DMF, 20 °C and 50% relative humidity. The scale bar is 10 μm. | ||
Just as observed previously for PVDF in DMF,15 and in line with the modeling results, the morphologies in Fig. 3a, c and d form due to water-vapor induced phase separation, initially giving polymer-rich droplets suspended in a DMF-swollen medium (e.g. depicted in Fig. 2b–e). During evaporation ripening and coalescence of the P(VDF-TrFE)-rich droplets take place until solvent is depleted and the film solidifies. The grain structure in the dry film suggests that at some point coarsening halts and the droplets impinge onto each other. The fact that DMF and anhydrous DMF give similar structure sizes indicates that water ingress is fast compared to solvent evaporation. VIPS seems to be virtually absent at 65 °C, even at 50% RH, which suggests (i) a decreased water miscibility at elevated temperature and/or (ii) suppression of VIPS domain growth due to fast solidification upon enhanced solvent evaporation. We note that a similar decrease in roughness with substrate temperature has also been observed for DMF-cast films of PVDF.15
Fig. 4 shows AFM topology images of P(VDF-TrFE) films cast from cyclohexanone (Fig. 4a and b) and cyclopentanone (Fig. 4c and d) at 20 °C, with the RH values varying between 10%, 50% and 80%. In excellent agreement with the model, microstructure formation due to VIPS is observed for cyclohexanone and not for cyclopentanone, even at 80% RH. It is however fair to note that the significantly lower vapor pressure of cyclohexanone (see the ESI†) further suppresses possible VIPS due to fast solidification of the film. The effect of solidification, which for P(VDF-TrFE) probably partly relies on crosslink formation via nano-crystallization, is not taken into account in our model.
 | ||
| Fig. 4 AFM height images of P(VDF-TrFE) thin films coated from cyclohexanone (a and b) and cyclopentanone (c and d). Conditions: (a and c) 20 °C, 50% RH, (b) 20 °C, 10% RH, (d) 20 °C, 80% RH. The scale bar is 10 μm. | ||
We note that the cyclohexanone used was not anhydrous. Hence, demixing might have been induced by water already present in solution prior to casting. This may also be the reason for the fact that similar structure sizes were obtained for different RH values, which contradicts the model prediction (Fig. 2b). In contrast, the predicted trend of an increasing structure size with decreasing humidity is in fact nicely reproduced in the older study on PVDF (Fig. 3a in that work).15 Just as for DMF, the cyclohexanone solutions produce smooth and uniform films at elevated substrate temperature (Fig. S4 of the ESI†). In the case of cyclopentanone, the surface roughness and morphology does not change substantially upon increasing the substrate temperature (see the ESI,† Fig. S5). Evidently, for ambient processing of P(VDF-TrFE), VIPS can only be avoided if a hydrophobic solvent with a water-miscibility well below 10 wt% is used.
In order to discriminate the VIPS-related dry film features from possible spherulitic crystallization, we also scanned the topology at the polymer–substrate interface. To this end, dry films were floated on the substrate and placed upside down under the AFM tip (see the Experimental section for details). The recorded images are given in Fig. 5 for cyclohexanone (Fig. 5a) and cyclopentanone (Fig. 5b). Since in both cases the topological and microstructural features are the same as those at the polymer–vapor interface heterogeneously nucleated spherulitic crystal growth can be ruled out, as that would not lead to topological gradients at the polymer–substrate interface.
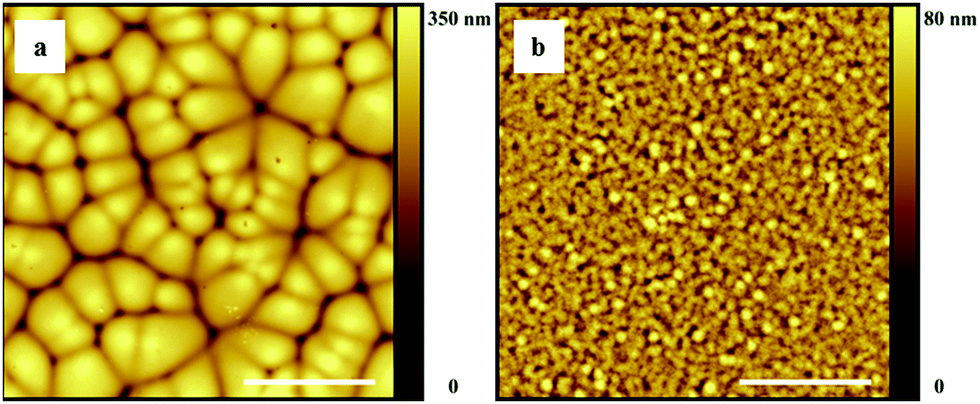 | ||
| Fig. 5 AFM topography images of the polymer/substrate interface of P(VDF-TrFE) layers cast from (a) cyclohexanone and (b) cyclopentanone. The substrate temperature and relative humidity were fixed at 20 °C and 50% in both cases. The scale bar is 10 μm. | ||
Film thickness and rms roughness acquired at 50% RH for all three solvents are plotted in Fig. 6 as a function of substrate temperature (also for 10% RH in Fig. S6, ESI†). Fig. 6a shows that for the films processed from DMF and cyclohexanone under ambient conditions the apparent mean thickness is strongly overestimated due to the large topological features resulting from VIPS (see also Fig. S7 in the ESI†). With increasing substrate temperature the structure densifies and the apparent mean thickness saturates at a value of 300 nm. Expectedly, the rms roughness follows the same trend (Fig. 6b). For cyclopentanone, however, due to the absence of VIPS, a dense smooth film of 300 nm with ∼10 nm rms roughness is already obtained at room temperature, irrespective of relative humidity.
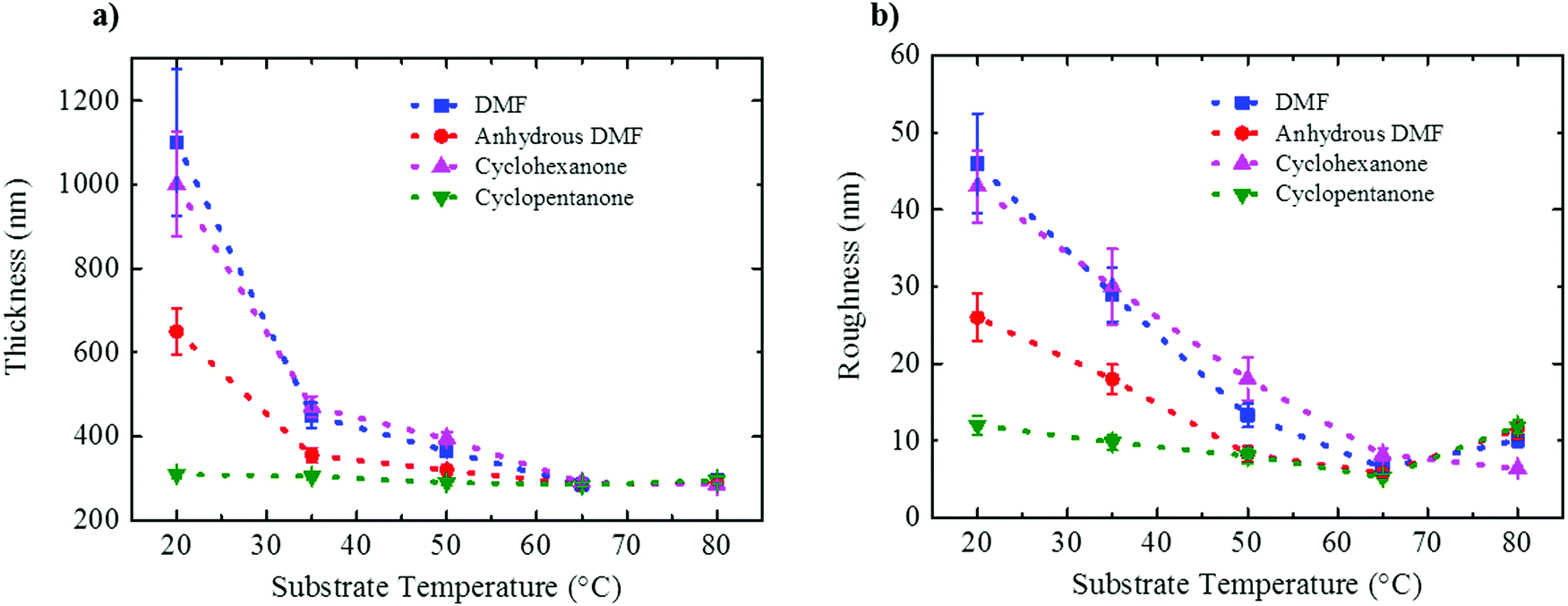 | ||
| Fig. 6 (a) Layer thickness and (b) rms roughness plotted as a function of substrate temperature measured for films prepared at a fixed relative humidity of 50%. | ||
Dry film morphology
In what follows we give our view on the origin of the impinged dry-film domain structure resulting from VIPS (e.g.Fig. 4a and b). The morphology suggests that at some point during drying coarsening ceases, whereby the polymer-rich droplets mutually impinge and deform from their circular or spherical shape. We speculate that elastic stresses occurring in the final stages of drying underlie this phenomenon. In general, free energy reduction by elastic deformation competes with domain coarsening driven by liquid–liquid interface minimization. The question is whether the dry film structure develops under the influence of purely elastic or viscoelastic stress. In the case of the former, domain deformation is caused by elastic asymmetry, such as a difference in shear modulus between the phases.33–35 Reduction of the total free energy is accomplished by anisotropic deformation of the softer phase into an extended network-like structure embedding domains of the harder phase.33In contrast, viscoeleastic structure development is driven by a dynamic asymmetry between the phases in a fluid mixture.36 In this case structure development is governed by the partitioning of the total stress between the phases, as governed by the force balance condition. In contrast to elastic demixing, morphologies develop in which the temporarily harder, i.e. more viscoelastic, phase forms the network surrounding domains of the “softer”/more fluid phase. We argue that the impinged dry film morphologies form under the influence of elastic rather than viscoelastic stress. We hypothesize that the elastic anisotropy, and the concomitant stagnation of coarsening, is introduced by late-stage development of P(VDF-TrFE) crystallites, giving rise to a gel-like state in polymer-rich regions.
Device characteristics
To probe the effect of solvent hygroscopicity on device performance we carried out a systematic study involving more than 100 ferroelectric capacitors fabricated per each solvent under ambient conditions (20 °C and 50% RH). The device architecture is depicted in the inset of Fig. 7b. The (apparent) mean film thickness was fixed at 〈d〉 = 150 ± 20 nm by adjusting the polymer concentration in solution. As shown by Fig. S8 in the ESI,† the morphology is not affected by varying the thickness. Enhanced crystallinity by subsequent annealing of the P(VDF-TrFE) films at 140 °C (see the Experimental section) only marginally increased the film roughness (Fig. S9 in the ESI†).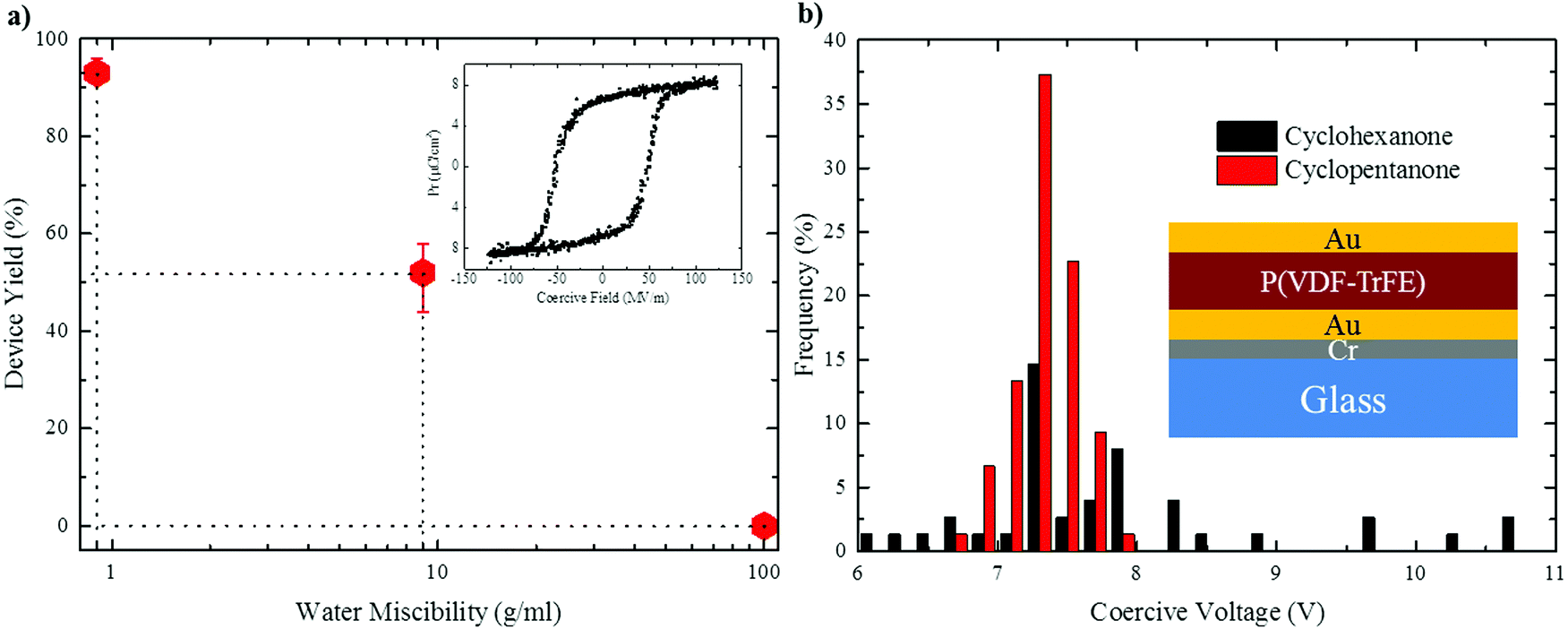 | ||
| Fig. 7 (a) Device yield as a function of water miscibility of solvents. (b) Histogram of coercive voltage for cyclohexanone and cyclopentanone. The insets show a ferroelectric displacement loop for the P(VDF-TrFE) capacitor and capacitor layout. The capacitors were measured with a continuous triangular bias at a frequency of 100 Hz and using a reference capacitor of 220 nF. | ||
A typical hysteresis loop of P(VDF-TrFE) is presented in the inset of Fig. 7a. The ferroelectric remanent polarization of all the capacitors was about the same and amounted to 6–7 μC cm−2, in agreement with literature data.3–5 The graph in Fig. 7a shows that the yield of functioning thin-film capacitors drops substantially with increasing water miscibility of the solvent: while no functional capacitor was obtained using DMF, cyclohexanone gave a yield of ∼50%. However, processing devices using cyclopentanone gave an impressive yield of >93%, the few non-working devices probably resulting from particle-induced short-circuits, rather than morphological flaws. Ferroelectric thin-film quality is also reflected by scatter in the coercive voltage, i.e. the voltage at which the polarization switches sign. Fig. 7b shows the histogram of the coercive voltages measured for capacitors fabricated from cyclohexanone and cyclopentanone. While the coercive voltage of cyclohexanone-processed devices shows significant scatter, a narrow distribution of 7.3 ± 0.5 V is obtained in the case of cyclopentanone, reflecting the low surface roughness and excellent film uniformity (ESI,† Fig. S9b).
Conclusions
The effect of ambient humidity, temperature and solvent hygroscopicity on the microstructure of P(VDF-TrFE) thin-films applied in flexible memory devices has been investigated. Computational predictions using a combination of phase diagram calculations and structural evolution modeling provide detailed insight into the mechanism of water vapor-induced phase separation (VIPS) during solution casting of the polymer under ambient conditions. Simultaneous solvent evaporation and water ingress destabilizes the blend in case the solvent is fully or partially miscible with water. We demonstrate that VIPS can occur even in solvents with water-miscibility as low as 10%. The calculations show that the time and length scales associated with the initial stages of VIPS exhibit an opposite trend with relative humidity. The model predicts the formation of polymer-rich droplets suspended in a solvent-rich medium, which is consistent with AFM analysis on solid P(VDF-TrFE) films cast from DMF and cyclohexanone, which indeed comprises clear drop-like features. In contrast, the more hydrophobic cyclopentanone gave smooth, near featureless layers, also in agreement with model predictions. Film quality is directly reflected by the thin-film capacitor yield which for DMF and cyclohexanone remains well below 50% with considerable scattering in coercive voltages, but increases to >93% and narrowly distributed coercive voltage when cyclopentanone is used.Author contributions
K. A. designed the experiment and supervised the work. H. S. D performed the experiments. J. J. M. performed the calculations. All the authors co-wrote the manuscript. All the authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to acknowledge Prof. P. W. M. Blom, Prof. D. M. de Leeuw and Dr M. Li for fruitful discussions, and Frank Keller, Verona Maus, and Thomas Lenz for their technical help. K. A. acknowledges the Alexander von Humboldt Foundation for funding provided in the framework of the Sofja Kovalevskaja Award endowed by the Federal Ministry of Education and Research, Germany. The authors acknowledge the financial support from the Max-Planck Institute for Polymer Research (Mainz, Germany). Open Access funding provided by the Max Planck Society.References
- R. C. Naber, K. Asadi, P. W. Blom, D. M. de Leeuw and B. de Boer, Adv. Mater., 2010, 22, 933–945 CrossRef CAS PubMed.
- P. Heremans, G. H. Gelinck, R. Muller, K.-J. Baeg, D.-Y. Kim and Y.-Y. Noh, Chem. Mater., 2010, 23, 341–358 CrossRef.
- A. J. Lovinger, Science, 1983, 220, 1115–1121 CAS.
- T. Furukawa, Adv. Colloid Interface Sci., 1997, 71, 183–208 CrossRef.
- D. Mao, M. Quevedo-Lopez, H. Stiegler, B. E. Gnade and H. N. Alshareef, Org. Electron., 2010, 11, 925–932 CrossRef CAS.
- R. Naber, P. Blom, A. Marsman and D. De Leeuw, Appl. Phys. Lett., 2004, 85, 2032–2034 CrossRef CAS.
- T. Furukawa, T. Nakajima and Y. Takahashi, IEEE Trans. Dielectr. Electr. Insul., 2006, 13, 1120–1131 CAS.
- R. C. Naber, C. Tanase, P. W. Blom, G. H. Gelinck, A. W. Marsman, F. J. Touwslager, S. Setayesh and D. M. De Leeuw, Nat. Mater., 2005, 4, 243–248 CrossRef CAS.
- M. A. Khan, U. S. Bhansali and H. N. Alshareef, Adv. Mater., 2012, 24, 2165–2170 CrossRef CAS PubMed.
- K. Asadi, D. M. De Leeuw, B. De Boer and P. W. Blom, Nat. Mater., 2008, 7, 547–550 CrossRef CAS PubMed.
- M. Li, N. Stingelin, J. J. Michels, M. J. Spijkman, K. Asadi, R. Beerends, F. Biscarini, P. W. Blom and D. M. de Leeuw, Adv. Funct. Mater., 2012, 22, 2750–2757 CrossRef CAS.
- A. Van Breemen, B. Kam, B. Cobb, F. G. Rodriguez, G. Van Heck, K. Myny, A. Marrani, V. Vinciguerra and G. Gelinck, Org. Electron., 2013, 14, 1966–1971 CrossRef CAS.
- A. J. van Breemen, J.-L. van der Steen, G. van Heck, R. Wang, V. Khikhlovskyi, M. Kemerink and G. H. Gelinck, Appl. Phys. Express, 2014, 7, 031602 CrossRef.
- H. Matsuyama, M. Teramoto, R. Nakatani and T. Maki, J. Appl. Polym. Sci., 1999, 74, 171–178 CrossRef CAS.
- M. Li, I. Katsouras, C. Piliego, G. Glasser, I. Lieberwirth, P. W. Blom and D. M. de Leeuw, J. Mater. Chem. C, 2013, 1, 7695–7702 RSC.
- M. Benz, W. B. Euler and O. J. Gregory, Macromolecules, 2002, 35, 2682–2688 CrossRef CAS.
- S. J. Kang, Y. J. Park, J. Hwang, H. J. Jeong, J. S. Lee, K. J. Kim, H. C. Kim, J. Huh and C. Park, Adv. Mater., 2007, 19, 581–586 CrossRef CAS.
- S. J. Kang, Y. J. Park, J. Sung, P. S. Jo, C. Park, K. J. Kim and B. O. Cho, Appl. Phys. Lett., 2008, 92, 012921 CrossRef.
- S. Ramasundaram, S. Yoon, K. J. Kim, J. S. Lee and C. Park, Macromol. Chem. Phys., 2009, 210, 951–960 CrossRef CAS.
- T. Lenz, H. Sharifi Dehsari, K. Asadi, P. W. Blom, W. A. Groen and D. M. de Leeuw, Appl. Phys. Lett., 2016, 109, 133302 CrossRef.
- PubChem Compound Database, http://https://pubchem.ncbi.nlm.nih.gov/compound/6228, accessed March 30, 2017.
- PubChem Compound Database, http://https://pubchem.ncbi.nlm.nih.gov/compound/7967, accessed March 30, 2017.
- PubChem Compound Database, http://https://pubchem.ncbi.nlm.nih.gov/compound/8452. accessed March 30, 2017.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, 1953 Search PubMed.
- C. Schaefer, J. J. Michels and P. van der Schoot, Macromolecules, 2016, 49, 6858–6870 CrossRef CAS.
- A. Ghodsi, H. Fashandi, M. Zarrebini, M. M. Abolhasani and M. Gorji, RSC Adv., 2015, 5, 92234–92253 RSC.
- H. Matsuyama, M. Teramoto, R. Nakatani and T. Maki, J. Appl. Polym. Sci., 1999, 74, 159–170 CrossRef CAS.
- J. J. Michels and E. Moons, Macromolecules, 2013, 46, 8693–8701 CrossRef CAS.
- C. Schaefer, P. van der Schoot and J. Michels, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 91, 022602 CrossRef CAS PubMed.
- A. Sariban and K. Binder, Macromolecules, 1988, 21, 711–726 CrossRef CAS.
- Since Fig. 5b represents a zoomed representation of the total phase diagram, only the binodal composition of the water-rich phase is depicted. The composition of the polymer-rich phase is obtained upon extrapolating the lower dashed lines to the other branch of the binodal curve.
- Y. Yip and A. J. McHugh, J. Membr. Sci., 2006, 271, 163–176 CrossRef CAS.
- A. Onuki and H. Nishimori, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 13649 CrossRef.
- C. Sagui, A. Somoza and R. C. Desai, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1994, 50, 4865 CrossRef CAS.
- D. Orlikowski, C. Sagui, A. Somoza and C. Roland, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 8646 CrossRef CAS.
- H. Tanaka, J. Phys.: Condens. Matter, 2000, 12, R207 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tc01495c |
| This journal is © The Royal Society of Chemistry 2017 |