
Polymeric electrochromic materials with donor–acceptor structures
Xiaojing
Lv
a,
Weijun
Li
a,
Mi
Ouyang
a,
Yujian
Zhang
 ab,
Dominic S.
Wright
*ac and
Cheng
Zhang
*a
ab,
Dominic S.
Wright
*ac and
Cheng
Zhang
*a
aCollege of Chemical Engineering, Zhejiang University of Technology, Chaowang Road No. 18, Hangzhou 310014, P. R. China. E-mail: czhang@zjut.edu.cn; Tel: +86-57188320508
bDepartment of Materials Chemistry, Huzhou University, Xueshi Road No. 1, Huzhou 313000, P. R. China
cDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: dsw1000@cam.ac.uk; Tel: +44 (0)1223763122
First published on 22nd November 2016
Abstract
Conjugated polymers with various electron-donor (D) and -acceptor (A) structures have been an important focus in the field of electrochromic (EC) research. Recent years have witnessed significant advances in the context of the design and synthesis of D–A type conjugated polymers. Most studies have investigated tunable band gap and color changes by introducing appropriate D and A units. However, D–A polymers with specific D units containing A units in the backbone or side chain possess varied ionization potentials, electron affinities and conjugation effects, leading to diverse electrochemical, optical-physical and EC properties. In addition, some innovative D–A structural polymers, such as cruciform and dendritic structures, present superior EC properties as well as multifunctional performance. In this review, our main focus will be placed on summarizing the characteristics of polymeric EC materials with various donor–acceptor structures. The overarching aim is to strengthen the understanding of the relationship between the D–A structure and the EC properties, especially color characteristics, and to provide some suggestions for the design of novel multifunctional D–A polymers for the future.
 Xiaojing Lv | Dr Xiaojing Lv received her PhD degree in Applied Chemistry from the Institute of Chemical Engineering and Technology at Zhejiang University of Technology in 2014. Then she conducted her postdoctoral research at Zhejiang University of Technology until now. Her current research focuses on the synthesis and application of novel electrochromic conjugated polymers. |
 Weijun Li | Dr Weijun Li received his PhD degree in Polymer Chemistry and Physics at the Jilin University in 2013. Then he underwent one year of postdoc research work at the University of Hong Kong. In 2015, he joined the College of Chemical Engineering, Zhejiang University of Technology. His current interest focuses on the synthesis and photophysical properties of organic photoelectronic materials, as well as their applications in the field of organic electrochromism, electroluminescence, and field effect transistors. |
 Mi Ouyang | Dr Mi Ouyang obtained his PhD in materials science at the Department of Polymer Science, Zhejiang University in 2008. Then he worked in the College of Chemical Engineering, Zhejiang University of Technology as an associate professor. His current research interests focus on the organic optoelectronic functional materials, including electrochromic and mechanochromic materials as well as organic–inorganic nanocomposites. |
 Yujian Zhang | Dr Yujian Zhang received his PhD degree from Zhejiang University of Technology (ZJUT) in 2012 under the supervision of Prof. Cheng Zhang. Now he is an Associate Professor in Huzhou University. His research interests include the development of external stimuli responsive molecular systems for molecular switches and chemosensors. |
 Dominic S. Wright | Prof. Dominic S. Wright received his PhD degree on lithium battery materials at the University of Cambridge in 1989. Then he obtained a research fellowship in Gonville and Caius College Cambridge. In 1991, he was appointed as a Lecturer in Inorganic Chemistry at The University of Cambridge before being promoted to his current position as Personal Chair in Inorganic Chemistry. In 2016, he joined Zhejiang University of Technology as an innovative professor. His research interests mainly focus on inorganic synthesis and materials chemistry, including macrocyclic main group compounds and energy storage materials. |
 Cheng Zhang | Prof. Cheng Zhang received his PhD degree in polymer materials at the Zhejiang University in 1998. In the following year, he did research work at Tokyo Institute of Technology as a JSPS postdoctoral fellow. In 2002, he joined College of Chemical Engineering, Zhejiang University of Technology as a professor. His main interest focuses on the design and synthesis of novel conjugated polymer materials, as well as their fundamental and application research in the field of organic optoelectronics and energy storage. |
1. Introduction
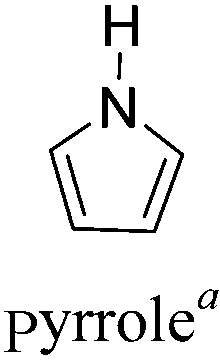
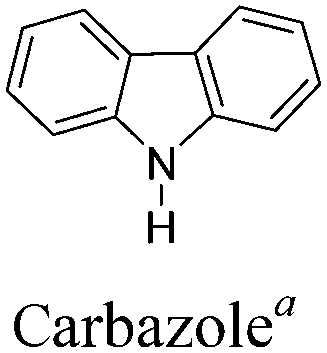
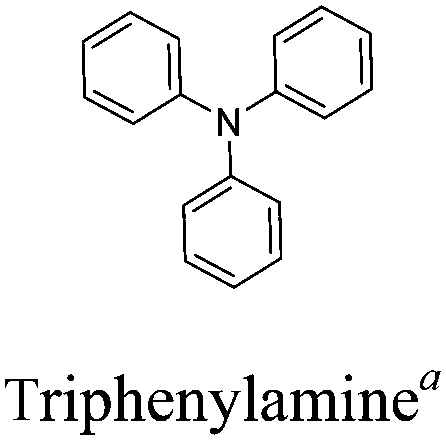
Conjugated polymers have gained popularity as a new generation of electrochromic (EC) materials due to their excellent processability, diverse colors, high optical contrast and fast switching properties.1–5 These valuable properties render them promising candidates for application in smart windows, reflectance mirrors, displays and military camouflage devices.6–9 In the last two decades, increasing research efforts have been devoted to the design and synthesis of polymeric EC materials possessing multicolored characteristics (including the three primary colors). It is widely believed that electrochromism in conjugated polymers occurs through changes in the π-electronic character of the polymer backbone, accompanied by reversible insertion/extraction of counterions in the electrolyte upon electrochemical oxidation and reduction.10 The neutral polymer shows semiconducting behaviour, having an energy gap Eg between the valence band (HOMO) and the conduction band (LUMO). Upon electrochemical doping, the band structure of the neutral polymer is changed, generating charge carriers (polarons or bipolarons) and lower energy intra-band transitions, which leads to different colors in both the neutral and doped forms. Therefore, one can vary the accessible colors through controlling the band gap of the conjugated polymers in both the neutral and doped materials.Among the various methodologies, the donor–acceptor (D–A) method developed by E. E. Havinga11 is the most efficient way to achieve tunable band gap polymers by alternation of D and A units in the polymer. Utilization of an appropriate choice of the D and A moieties allows selection of the approximate HOMO and LUMO energies of the resulting polymers, leading to a fine control of the band gap, optical character and EC properties.12–16 To date, many groups have reported D–A type conjugated polymers with tunable band gaps and considerable EC properties.17–30 Most of the D–A polymeric materials have been investigated for the electrochromic behaviors in their oxidized states under a positive applied potential, due to the instability of the reduced polymers upon exposure to air. Therefore, the D unit plays a crucial role in the D–A type conjugated polymers, which determines the structure of the polymer, optical character, fundamental colors and other EC properties. Conventional D units include thiophene, EDOT, pyrrole, triphenylamine, carbazole and their derivatives,31–38 and their HOMO and LUMO values are given in Table 1.39–44 However, previous reports have mainly investigated the influence of the band gap on the color changes of the conjugated polymers through changing of D or A units, and there has been little systematic discussion about the effect of various D–A structures on the EC properties. Hence, this review article focuses on D–A type conjugated polymers bearing specific D units and A units in their backbones, and their effects on the EC properties. In addition, some innovative D–A structural polymers, including pendent A units, and cruciform and dendritic D–A structures, are also involved in this review. The overall aim is to strengthen the understanding of the relationship between the D–A polymeric structure and the EC properties, especially color characteristics, and provide some pointers to new multifunctional polymeric EC materials.
| Donor unit | HOMO (eV) | LUMO (eV) | E g (eV) |
|---|---|---|---|
| a Data taken from ref. 39–44. | |||

|
−6.486 | −0.408 | 6.078 |

|
−5.81 | −0.06 | 5.75 |
|
−5.60 | 1.25 | 6.85 |
|
−5.44 | −0.64 | 5.38 |
|
−4.95 | −0.30 | 4.65 |
2. EC polymers with D and A units in the backbone
2.1 Thiophene and derivatives as D units
Polythiophenes have been most widely studied as promising EC materials due to their excellent stability and easy tailoring of the thiophene monomers, which can be switched between neutral red color (λmax = 470 nm) with a band gap of 1.94 eV and blue color (polaron absorption at 730 nm) in the doped state.45 A large amount of work has been reported on D–A polymers bearing thiophene as D units with tunable colors through the introduction of different electron-acceptor units (in Table 2). For instance, A. Erden et al.46 reported a D–A polymer P1 containing benzothiadiazole as the A unit, which exhibited a neutral purple-red color with absorption at about 560 nm and a band gap of 1.5 eV. Upon doping, it switched to a blue color due to two new peaks at about 760 nm (polarons) and 1100 nm (bi-polarons). P2 containing benzooxadiazole as the A unit, prepared by A. M. Önal and A. Cihaner et al.47 showed magenta color in the neutral state with a band gap of 1.48 eV, which switched to light navy blue upon oxidation. Likewise, by introducing different acceptor units, P3,48P449 and P550 (Table 2) possess bandgaps of 1.65 eV, 1.55 eV and 1.91 eV, respectively. All the polymers showed neutral purple or red colors and switched to a blue color upon oxidation. Collectively, these D–A polymers show band gaps in the range of 1.4 to 2.0 eV and their absorption bands are very close to the parent polythiophene, hence they have similar neutral and oxidized colors to polythiophene. While D–A polymers achieve a higher band gap (>2.0 eV), they may exhibit different neutral colors from polymers with the band gaps in the range of 1.4 to 2.0 eV, remaining a similar blue color in the oxidized state. For instance, P6, P7 and P8 synthesized by L. Toppare and Y. A. Udum et al.49,51,52 (Table 2) exhibit neutral maximum absorption bands at 490 nm, 430 nm and 390 nm with the band gaps of 2.0 eV, 2.2 eV and 2.25 eV, respectively. Upon oxidation, the polymer films altered their colors from yellow to grey or blue, due to new polaron and bipolaron bands. Therefore, when the band gaps of D–A polymers bearing thiophene units reach more than 1.4 eV, the introduction of A units can only change the neutral color of the polymer and has less effect on their oxidized colors. In contrast, D units have a more important role in the oxidized colors of the polymers.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| A | R1 | R2 | λ abs (nm) | E g (eV) | Neutral color | Oxidized color | Ref. | |
| a Absorption maximum of the neutral-state polymer film. b Optical band-gaps as calculated from the onset of the π–π* transition of the neutral-state polymer. | ||||||||
| P1 |

|
–H | –H | 560 | 1.5 | Purple | Blue | 46 |
| P2 |

|
–H | –H | 345, 560 | 1.48 | Magenta | Light navy | 47 |
| P3 |

|
–H | –H | 558 | 1.65 | Purple | Blue | 48 |
| P4 |
|
–H | –H | 504 | 1.55 | Red | Blue | 49 |
| P5 |
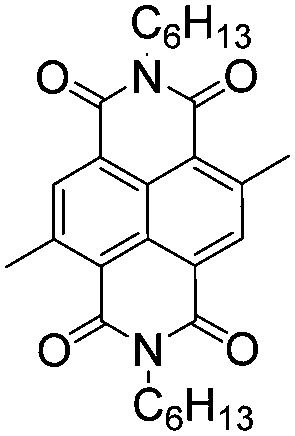
|
–H | –H | 500 | 1.91 | Red | Blue | 50 |
| P6 |
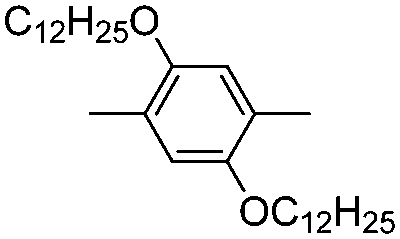
|
–H | –H | 490 | 2.0 | Yellow | Blue | 51 |
| P7 |
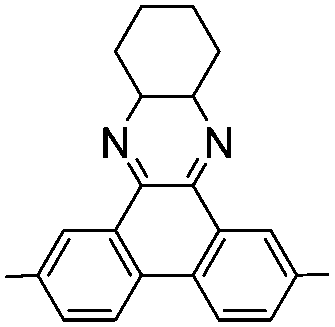
|
–H | –H | 395 | 2.2 | Yellow | Blue | 52 |
| P8 |

|
–H | –H | 390 | 2.25 | Yellow | Gray | 49 |
| P9 |

|
–H | –H | 370, 725 | 1.10 | Green | Transmissive brown | 54 |
| P10 |

|
–H | –H | 450, 950 | 1.02 | Green | Transparent | 55 |
| P11 |

|
–H | –H | 432, 1020 | 0.98 | Green | Transparent | 55 |
| P12 |

|
–H | –H | 400, 886 | 0.92 | Green | Transparent | 55 |
| P13 |
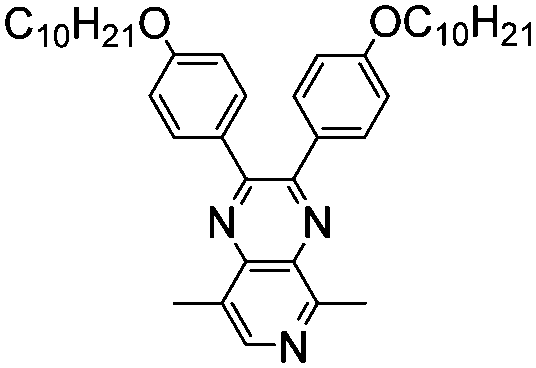
|
–H | –CH3 | 398, 700 | 1.39 | Light blue | Colorless | 56 |
| P14 |

|
–H | –OCH3 | 408, 820 | 1.136 | Green | Transmissive grey | 56 |
| P15 |

|
–OCH3 | –OCH3 | 382, 657 | 1.25 | Light blue | Transmissive brown | 56 |

Introducing stronger electron-withdrawing groups into D–A polymers bearing thiophene units may result in narrow band gaps53 in the range of 0.5–1.4 eV, and more interesting EC behavior. F. Wudl et al.54 reported a novel D–A polymer P9 containing thienopyrazine as the A unit, which showed a narrow band gap of 1.10 eV and was demonstrated to be the first neutral green polymer. The unique green color was mainly attributed to the two main absorption bands in the visible region at 370 nm and 725 nm. Upon oxidation, the polymer film switched to a transmissive brown. The polymer with green color properties makes it promising to realize full-color displays. After this study, a range of other works have reported green neutral polymers. L. Toppare et al.55 synthesized several D–A polymers P10–P12 with the benzotriazole substituted quinoxalines as the A units. All the polymers had low band gaps (ca. 1.00 eV) and a green color in the neutral state, which switched to transparent in their oxidized states owing to the formation of polaron and bipolaron in the NIR region. P13–15 (Table 2), containing 3-methyl-, 3-methoxyl-, 3,4-dimethoxyl-substituted thiophene as D units and pyrido[3,4-b] pyrazine as A units, prepared by J. S. Zhao and M. Wang et al.56 also exhibited the low band gap of 1.39 eV, 1.13 eV and 1.25 eV, respectively. As a result, they exhibited blue or green color in the neutral state, and switched to being almost colorless and highly transmissive in their oxidized states. Consequently, D–A polymers bearing thiophene with highly narrow band gaps (<1.4 eV) can exhibit a neutral green or blue color through the introduction of appropriate A units into the backbone. These polymers might have displayed the transmissive state because of the shift to the longer NIR wavelength upon oxidation.
2.2 EDOT and derivatives as D units
As the star derivative of polythiophene, poly(3,4-ethylenedioxythiophene) (PEDOT) can be switched between a neutral dark blue, with a band gap of 1.6 eV (the absorption band at about 620 nm), and light blue in the oxidized form. The rapid switching speed, low oxidation potential and good stability make it useful for the synthesis of advanced EC materials.57–60 Much work has been reported on the design and synthesis of D–A polymers bearing electron-rich EDOT in the backbone (in Table 3). For example, L. Toppare et al.61,62 synthesized several novel D–A polymers P16–P18 bearing EDOT as the donor, and benzotriazole and benzimidazole as the acceptor units (Table 3). These polymers showed the maximum absorption at about 618 nm, 580 nm and 513 nm with the band gaps of 1.6 eV, 1.75 eV, 1.77 eV, respectively, which were very close to that of the parent PEDOT. Hence, they exhibited neutral blue or purple color and turned transparent blue in the oxidized states. Increasing the band gap to more than 1.9 eV through the introduction of weaker electron-withdrawing units into D–A polymers provides the potential for tunable neutral electrochromism with the oxidized blue color. P19 synthesized by J. R. Reynolds et al.63,64 containing pyridine as the A unit exhibited a red neutral state with the band gap of 1.9 eV and blue-purple in the oxidized state. With methoxy-ethylhexyloxy-benzene as the A unit,65 the polymer P20 possesses a neutral absorption band at 535 nm with a high band gap of 1.95 eV, giving a very saturated purple-red color, which turned into deep-blue upon oxidation. P21 and P22 showed orange or yellow color with neutral absorption bands at 490 nm and 357 nm and with the band gaps of 2.0 eV and 2.66 eV, respectively, switched to a blue color in the oxidative state.52,66 Hence, the neutral color of D–A polymers with EDOT as the donor unit can be adjusted by changing the electron-withdrawing units in the backbone. The oxidized blue or transparent blue color can be pre-determined by utilising the EDOT unit, with a band gap of more than 1.6 eV.|
|
||||||
|---|---|---|---|---|---|---|
| A | λ abs (nm) | E g (eV) | Neutral color | Oxidized color | Ref. | |
| a Absorption maximum of the neutral-state polymer film. b Optical band-gaps as calculated from the onset of the π–π* transition of the neutral-state polymer. | ||||||
| P16 |
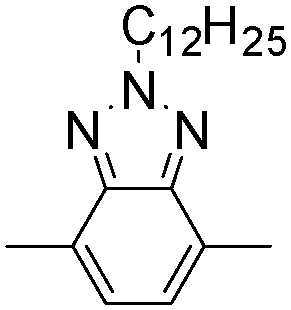
|
618 | 1.6 | Blue | Transparent blue | 61 |
| P17 |
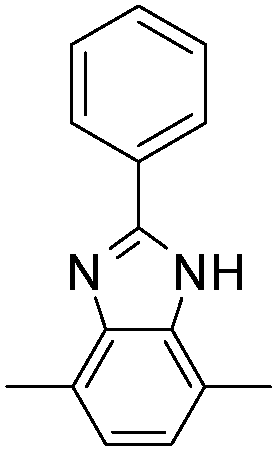
|
580 | 1.75 | Blue | Transparent blue | 62 |
| P18 |

|
513 | 1.77 | Purple | Blue | 62 |
| P19 |

|
478 | 1.9 | Red | Blue-purple | 63 and 64 |
| P20 |
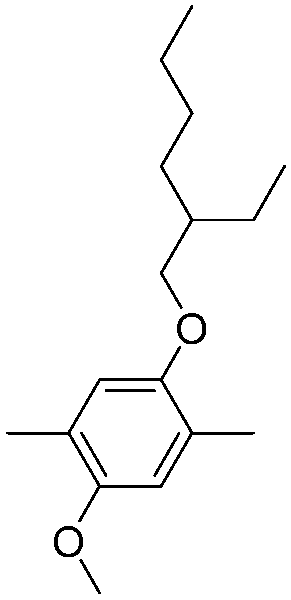
|
535 | 1.95 | Purple-red | Dark blue | 65 |
| P21 |

|
490 | 2.0 | Orange | Blue | 52 |
| P22 |

|
357 | 2.66 | Yellow | Blue | 66 |
| P23 |
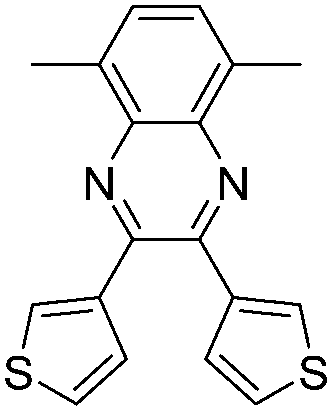
|
405, 780 | 1.2 | Green | Transparent | 67 |
| P24 |

|
452, 711 | 1.18 | Green | Colorless | 68 |
| P25 |
|
415, 690 | 1.45 | Green | Transparent | 69 |
| P26 |
|
427, 746 | 1.25 | Green | Transmissive blue | 70 |
| P27 |

|
428, 755 | 1.19 | Green | Transmissive light blue | 71 |

While D–A polymers with lower band gaps (<1.6 eV) than pristine PEDOT might possess different EC behaviours compared to the above mentioned EDOT counterparts, L. Toppare et al.67–69 reported several D–A polymers P23–P25 containing quinoxaline as the A unit, with band gaps of about 1.4 eV. All the polymers revealed two maximum absorption bands at around 400 and 700 mm in their visible region, thus exhibiting a green color in the neutral state, with a highly transmissive oxidized state. P26 with pyrrole-quinoxaline as the A unit synthesized by F. Algı and A. Cihaner70 revealed a highly stable neutral state green polymer. The polymer exhibited three well-defined absorption bands with a narrow band gap (1.25 eV), and turned a highly transmissive blue color in the oxidized state. Similarly, P27 bearing benzothiadiazole as the A unit also presented two well separated absorption maxima at 428 nm and 755 nm with a low band gap of 1.19 eV. As a result, it exhibited a green color in the neutral state and a highly transmissive light blue color in the oxidized state.71 Therefore, the introduction of appropriate A units can control the band gap of D–A polymers containing EDOT to less than 1.6 eV, and realize a green color in the neutral state and a transmittance oxidative state.
As the most promising derivative of PEDOT, alkyl-or alkoxy-substituted poly(3,4-propylenedioxythiophene)s (PProDOT)s, first reported by J. R. Reynolds et al.,72–74 possess high-lying HOMO energy levels which were capable of switching between a blue neutral state and a highly transmissive sky-blue oxidized state in sub-second time frames, as well as high optical switching contrast ratios and high coloration efficiencies. Following this work, A. M. Önal and A. Cihaner et al.75,76 reported a series of D–A type polymers P28–P31 (Table 4) containing dodecyl substituted ProDOT as the D units and benzoselenadiazole, benzooxadiazole, benzothiadiazole, and benzotriazole as the A units. It was found that P28, P29, and P30 had similar optical characteristics, bearing two absorption bands at about 400 nm and 700 nm and with band gaps of around 1.40 eV, exhibiting green, cyan and greenish-blue colors in their neutral states, respectively. Upon oxidation, all polymer films exhibited highly transmissive colors. Since a weak electron-withdrawing unit is present, P31 exhibits a relatively high band gap of 1.80 eV, close to the parent PEDOT, leading to a neutral blue color and a transparent oxidized state. More interestingly, all these polymers showed high optical contrast, coloration efficiencies and solubility in common organic solvents, which can be ascribed to the less dense morphology provided by long alkyl substituents, thus allowing for faster ion movement and more dopant ions during redox switching.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| A | R | λ abs (nm) | E g (eV) | Neutral color | Oxidized color | Ref. | |
| a Absorption maximum of the neutral-state polymer film. b Optical band-gaps as calculated from the onset of the π–π* transition of the neutral-state polymer. | |||||||
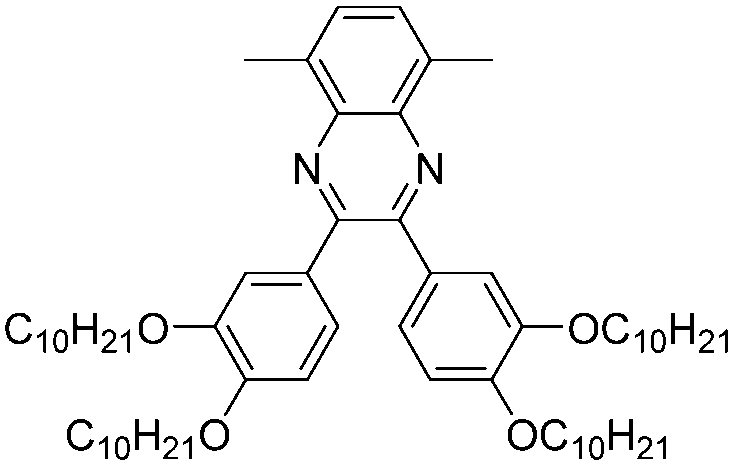
| P28 |
|
–C10H21 | 419, 700 | 1.37 | Green | Transparent | 75 |
| P29 |

|
–C10H21 | 400, 697 | 1.40 | Cyan | Transmissive grey | 76 |
| P30 |
|
–C10H21 | 408, 685 | 1.48 | Greenish-blue | Transparent | 76 |
| P31 |

|
–C10H21 | 585 | 1.80 | Blue | Transparent | 76 |

2.3 Pyrrole and derivatives as D units
Compared to polythiophene and PEDOT, polypyrrole has a relatively high band gap of 2.7 eV, which can be switched from neutral yellow to blue-violet in the doped state. Hence, there exists a little interest in the utilization of pyrrole as the donor unit to construct D–A polymers77–79 L. Toppare et al.80–82 reported several D–A polymers P32–P35 containing pyrrole as the D units, and quinoxaline, benzothiadiazole and benzoselenadiazole as the A units in the backbone (Table 5). All the polymers possess narrow band gaps of 1.10 eV with two distinct π–π* transition absorption bands at around 400 and 750 nm in the neutral state. Thus these polymers exhibited green color and switched to a bluish state upon oxidation. Hence, pyrrole-bearing D–A polymers can exhibit narrow band gaps (<1.4 eV) through the introduction of strong electron-withdrawing units into the backbone. The polymers are able to achieve a neutral green color, whereas their oxidized colors are still affected by the D unit and are similar to pristine polypyrrole.|
|
||||||
|---|---|---|---|---|---|---|
| A | λ abs (nm) | E g (eV) | Neutral color | Oxidized color | Ref. | |
| a Absorption maximum of the neutral-state polymer film. b Optical band-gaps as calculated from the onset of the π–π* transition of the neutral-state polymer. | ||||||
| P32 |

|
392, 753 | 1.12 | Green | Blue | 82 |
| P33 |

|
448, 796 | 1.13 | Green | Transmissive sky blue | 82 |
| P34 |

|
408, 745 | 1.25 | Green | Gray | 80 |
| P35 |
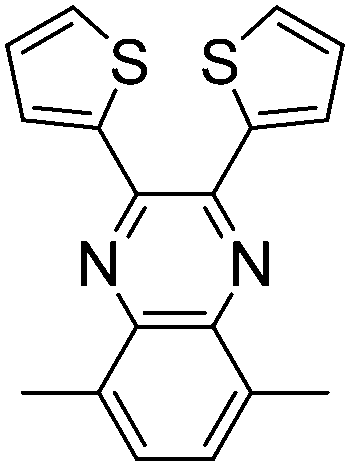
|
400, 815 | 1.0 | Green | Brownish green | 81 |

Due to the intrinsic structural character of pyrrole, the N-position of pyrrole can also be substituted by modified groups, providing novel D–A polymers with pyrrole units in the backbone and A units at the side chain. This type of polymer will be discussed in the next section in detail.
2.4 Carbazole and derivatives as D units
A carbazole monomer can be easily functionalized at its (3,6-), (2,7-) or N-positions and then covalently linked into polymeric systems. A 3,6-linked carbazole moiety in the main chain of the polymer serves as a conjugation break, which in turn leads to some useful EC properties. Upon initial oxidation, an electron is removed from the conjugated backbone and a radical cation is formed. Because of the conjugation breaks, the radical cations are separated from each other and cannot combine. Upon further oxidation, another electron is removed, giving dications. Hence, the ability to support two distinct oxidation states of 3,6-linked polycarbazole derivatives leads to multi-colored characteristics. For instance, J. R. Reynolds et al.83 synthesized a novel D–A polymer P36, containing 3,6-linked N-octylcarbazole as the D unit and benzothiadiazole as the A unit (Table 6). This polymer showed the greatest absorption at 460 nm in the neutral state with a broad band gap of 2.6 eV, showing an orange color. Due to the conjugation break effect, the polymer showed a green-grey color during the radical cation state, and switched to a grey color in its most oxidized state. Y. A. Udum et al.84 synthesized a D–A type polymer P37 bearing oxadiazole as the A moiety. Polymer films had no sharp absorption peak in the visible range in the neutral state with a band gap of 2.2 eV, giving a yellow color. Applying a potential, the polymer films gave absorption bands at 480 nm and 720 nm at 1.2 V, giving a green color and can be further switched to a blue color due to increased absorption at 600 nm. F. B. Koyuncu85 also reported one D–A polymer P38 bearing naphthalene bisimide as the A unit. The polymer film exhibited two reversible oxidation waves and the transparent film in the neutral state turned into green and then turquoise resulting from the association of polaronic and bipolaronic species from carbazolylium cation radicals.|
|
|||||||
|---|---|---|---|---|---|---|---|
| A | R | λ abs (nm) | E g (eV) | Neutral color | Oxidized color | Ref. | |
| a Absorption maximum of the neutral-state polymer film. b Optical band-gaps as calculated from the onset of the π–π* transition of the neutral-state polymer. | |||||||
| P36 |
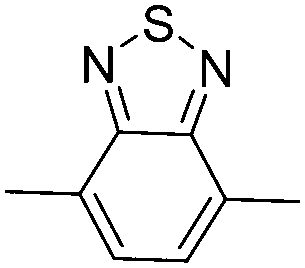
|
–C8H17 | 460 | 2.6 | Orange | Green-gray, gray | 83 |
| P37 |

|
–CH3 | — | 2.2 | Yellow | Green, blue | 84 |
| P38 |

|
–C2H5 | 360, 378 | 3.1 | Transparent | Yellowish green turquoise | 85 |

Due to the intrinsic structural character of carbazole, the N-position of carbazole can also be substituted by A units in the backbone in the construction of D–A polymers. And the substituent position of carbazole with A units may bring out different optical–physical characteristics. For instance, J. S. Zhao et al.86 obtained a polymer containing benzothiadiazole connected to N-substituted carbazole P39 (Fig. 1). Compared to P36, this polymer film exhibited an absorption band at 470 nm with a band gap of 2.26 eV due to the π–π* transition, giving a neutral yellow color. As the potential was increased, the absorption bands at 420 and 800 nm increased gradually, leading to a green color. Upon further oxidation, films turned turquoise blue. Therefore, the neutral and oxidized colors of D–A polymers can be tuned via the introduction of acceptor units on different substituent positions of the carbazole unit.
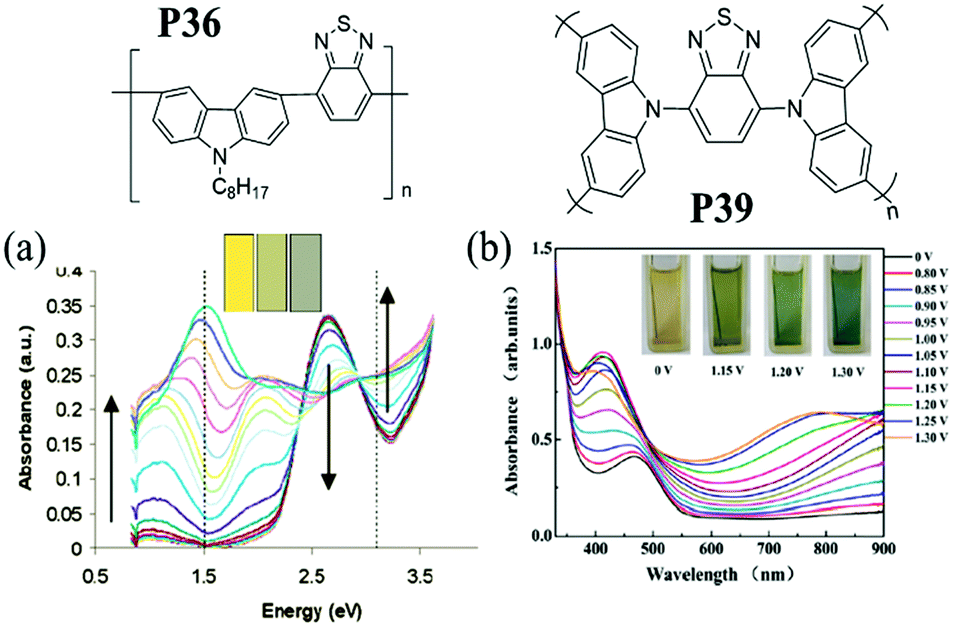 | ||
| Fig. 1 Molecular structure of P36 and P39, spectroelectro-chemical spectra and colors at different potentials. (a) Adapted with permission from ref. 83. Copyright 2005 American Chemical Society. (b) Adapted with permission from ref. 86. Copyright 2013 Elsevier. | ||
2.5 Randomly built-up D units
The previous sections have reviewed the EC character of different donor systems and emphasized that D units play a critical role in color control of D–A polymers in the doped state. Thereby, random combinations of two or more donor units in a D–A polymer with an appropriate A unit may create more intriguing electrochromism. J. R. Reynolds et al.87 reported a series of D–A polymers P40–P46 containing 3,4-bis(2-ethylhexyloxy)thiophene (DOT-(OEtHx)2) and 2-ethylhexyl substituted ProDOT (ProDOT-(CH2OEtHx)2) as the D units, and benzothiadiazole as the A unit. When the monomer P43 was copolymerized with four equivalents of ProDOT-(CH2OEtHx)2, the most surprising discovery was that the copolymer P46 possessed an absorption spectrum which extended over the entire visible region (400–700 nm) with a band gap of 1.64 eV, owing to the complementary absorption of P43 (at 396 nm, 644 nm) and PProDOT-(CH2OEtHx)2 (at 543 nm and 576 nm). Consequently, a neutral black polymer could be realized and switched to a highly transmissive oxidized state without absorption in the visible region (Fig. 2). This significant work makes the polymer desirable as an EC material for low-voltage smart windows. | ||
| Fig. 2 Molecular structure of P40–P46, spectroelectrochemical spectra and color showing between 0.04 V and 0.74 V of P46. Adapted with permission from ref. 87. Copyright 2008 Nature. | ||
In addition, Reynolds et al.88 investigated molecular structure effects, redox properties, and electrochromic performance of D–A polymers P47–P52 with random combinations of electron-rich 3,4-dioxythiophene (DOT) units, including thiophene, DOT-(OMe)2, DOT-(OEtHx)2 or ProDOT-(CH2OEtHx)2 and the electron-deficient core of benzothiadiazole (Fig. 3). All the polymers exhibited the dual band of optical absorption with a low band gap (∼1.6 eV) regardless of the type of constituting D units, resulting in a neutral green color and the transmissive state as oxidized. Random combinations of “unbridged” dialkoxythiophene (DalkOT) and ProDOT counterparts (P48–P52) exhibited a lower onset oxidation potential than the homopolymer bearing DalkOT units (P47). In addition, D–A polymers (P50–P52) bearing ProDOT building units yielded better optical contrast and long-term stability due to their higher HOMO value.
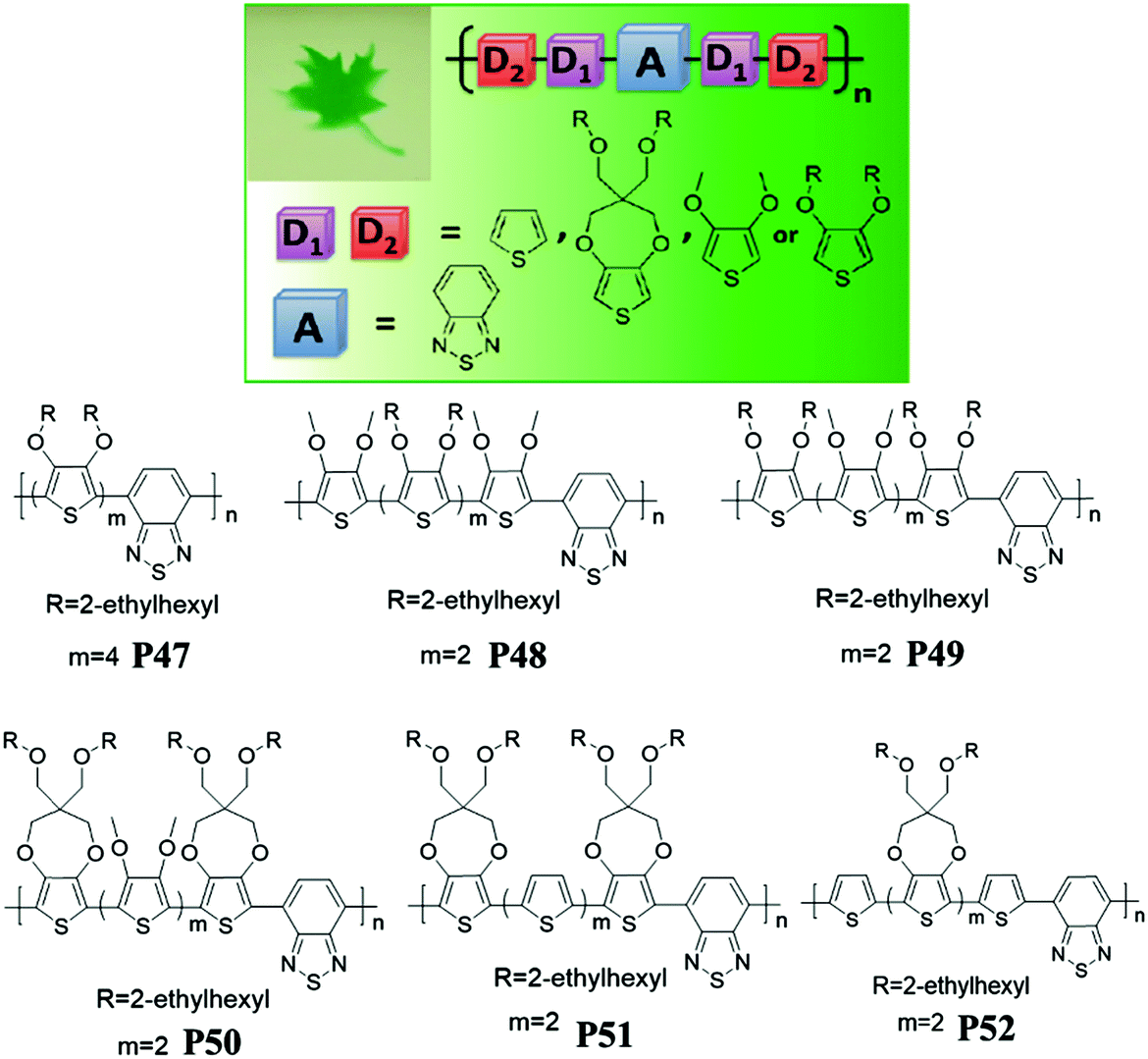 | ||
| Fig. 3 Molecular structures of P47–P52 with neutral green color shown (top left corner). Adapted with permission from ref. 88. Copyright 2012 American Chemical Society. | ||
J. W. Xu et al.89 have also done some work on the random D–A polymers. For instance, they synthesized P53–P56 combining DalkOT and EDOT as the donor units, and 2-alkylbenzotriazole as acceptor units. The polymer P55 film showed one strong absorption band at 590 nm, thus giving a saturated blue color (Fig. 4). As oxidized, it switched into a transparent color due to no absorption in the visible region, compared to P16 bearing single EDOT as the D unit with a light transparent blue color in the oxidized state. In addition, J. W. Xu and Y. Liu et al.90 also synthesized another random D–A polymer P57 with a combination of thiophene and carbazole units. The EC devices fabricated from spin-coated polymer films could switch reversibly between deep blue and transmissive light green hues, with a high optical contrast of 41% in the visible region and a long-term stability of over 7500 cycles under ambient conditions with limited reduction (Fig. 5). In conclusion, D–A polymers bearing randomly built D units can possess a combination of the EC characteristics for individual donor systems.
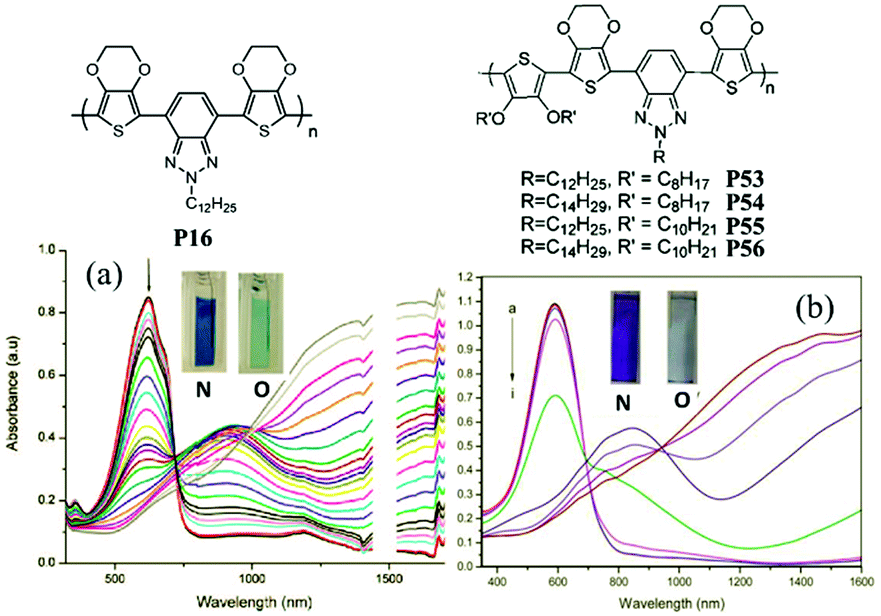 | ||
| Fig. 4 Molecular structure of P16, P53–P56; spectroelectro-chemical spectra and colors of P16 (a), P55 (b) in the neutral and oxidized states. Adapted with permission from ref. 61. Copyright 2008 American Chemical Society and ref. 89. Copyright 2013 The Royal Society of Chemistry. | ||
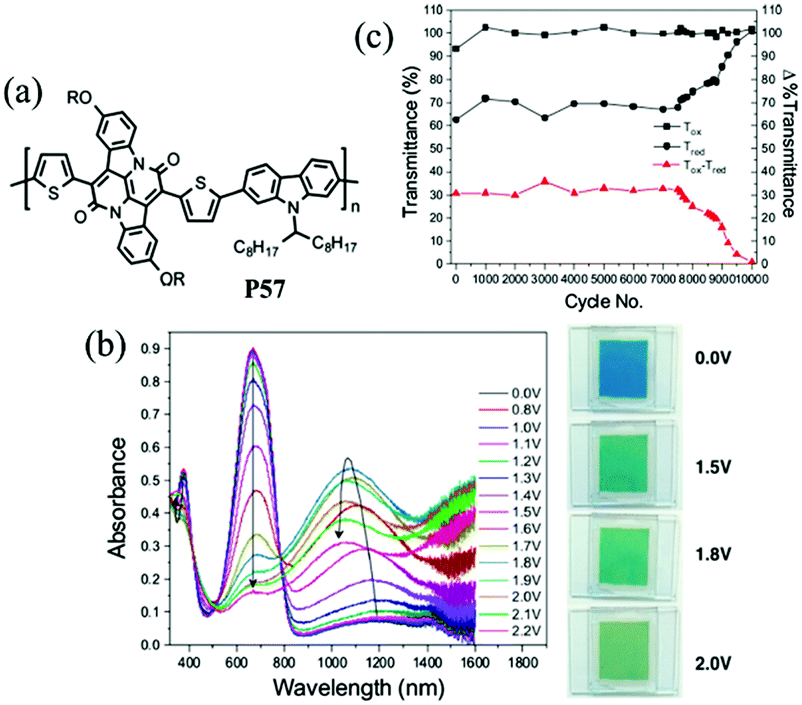 | ||
| Fig. 5 (a) Molecular structure of P57, (b) spectroelectro-chemical spectra and colors under different potentials, (c) long-term stability of EC devices. Adapted with permission from ref. 90. Copyright 2016 American Chemical Society. | ||
In order to investigate the effect of different donor groups on the optical properties of D–A type polymers further, an asymmetric polymer P58 (Fig. 6) was reported by L. Toppare et al.91 using the combination of EDOT and thiophene donor units. Compared with its symmetrical thiophene (P1) and EDOT (P27) relatives, the polymer exhibited two neutral absorption bands at 408 nm and 642 nm, which demonstrated a clear shift in the dominant wavelength as the co-monomer composition was varied from pristine P1 to P27 (Fig. 6). Correspondingly, this polymer showed an intermediate color between those of P1 and P27. Furthermore, it had a band gap of 1.18 eV, which was lower than any of the individual homologues, and also had superior EC properties to the copolymers synthesized using the monomers of P1 and P27.
 | ||
| Fig. 6 Normalized absorbance spectra of P1, P27, P58 and the copolymer films prepared from different compositions (all in the neutral state). Adapted with permission from ref. 91. Copyright 2010 Elsevier. | ||
3. EC polymers with D and A units in particular structures
3.1 D–A polymers with pendent A units
The N-positions of pyrrole or carbazole can be substituted by modified groups, providing novel D–A polymers with pyrrole or carbazole units in the backbone and A units at the side chain. For instance, Y. B. Shim et al.92 synthesized two D–A polymers P59 and P60 containing 2,5-di(2-thienyl)-1H-pyrrole (SNS) as the D unit, 3-pyridinyl and 1,10-phenanthroline as A units (Table 7). The polymer P59 presented a brownish-yellow color in the neutral state (at 430 nm) with a band gap of 2.1 eV and was dark blue in the fully oxidized state while P60 showed a greenish-yellow color in the neutral state (at 451 nm) with a lower band gap of 1.85 eV and was light blue in the fully oxidized state. These polymer films exhibited a fast switching time of within 1.0 s. A. Cihaner et al.93 reported a D–A polymer P61 (Table 7) containing 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) as the A unit. The polymer film showed a neutral purple color at 351 nm with a band gap of 2.9 eV. Upon oxidation, the polymer film switched to violet, then to grey blue. Compared to PSNS, this polymer with BODIPY at the side chain had lower response time for switching between the neutral and oxidized states (within 1.0 s) and higher electroactive stability. Consequently, D–A polymers containing pyrrole as a D unit with pendent A units possess relatively smaller color changes both in the neutral and oxidized states than those containing A units in the backbone. However, the pendent structural D–A polymers commonly show relatively fast switching time (within 1 s) compared to most of the linear D–A polymers under the other similar conditions (such as electrode, interfacial properties and film thickness).|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| A | λ abs (nm) | E g (eV) | Neutral color | Oxidized color | Switching timec (s) | Optical contrastc (%) | Ref. | |
| a Absorption maximum of the neutral-state polymer film. b Optical band-gaps as calculated from the onset of the π–π* transition of the neutral-state polymer. c Switching time and optical contrast in the visible region. | ||||||||
| P59 |
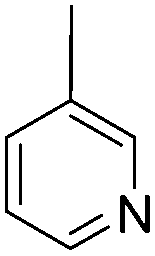
|
430 | 2.1 | Brownish-yellow | Dark blue | 1.3 | 42 | 92 |
| P60 |

|
451 | 1.85 | Greenish-yellow | Light blue | 0.92 | 31 | 92 |
| P61 |
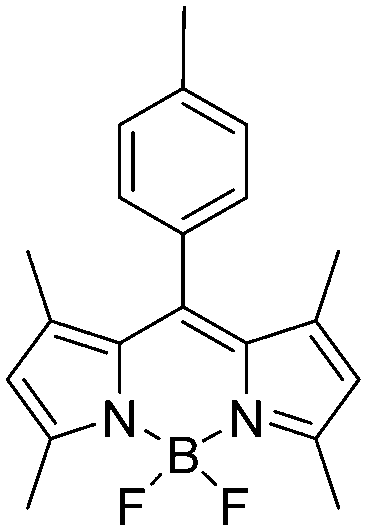
|
351 | 2.9 | Purple | Grey blue | 1.0 | 28.6 | 93 |

Our group has researched the EC behaviour of N-substituted polycarbazole with pendent A units. A series of D–A polymers P62–P64 (Table 8) containing 2,5-di(2-thienyl)-1H-carbazole as the D unit and with ethyl-phenylmethanone and nitryl as the A units at the N-position have been investigated.94,95 All the three polymers exhibited a neutral absorption band at about 415 nm with a yellow color and the band gaps were estimated to be more than 2.0 eV. Upon further oxidation, the increased absorption at 600 nm together with a diminished neutral state absorption resulted in a blue or blue-purple color. Hence, it has also been shown that D–A polymers containing carbazole as the D unit with different pendent A units possess a relatively high band gap (∼2 eV), and have little effect on the electrochromism in the neutral and oxidized states. Nevertheless, all these polymers showed a high coloration efficiency of about 200 cm2 C−1.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| A | λ abs (nm) | E g (eV) | Neutral color | Oxidized color | Coloration efficiencyc | Ref. | |
| a Absorption maximum of the neutral-state polymer film. b Optical band-gaps as calculated from the onset of the π–π* transition of the neutral-state polymer. c Coloration efficiency in the NIR region. | |||||||
| P62 |
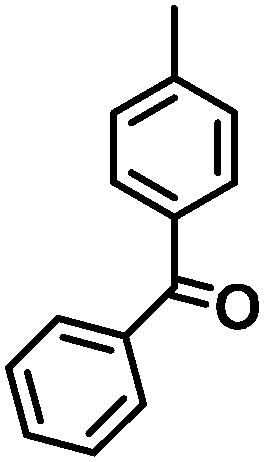
|
415 | 2.3 | Yellow | Brownish-yellow | 186.18 | 95 |
| P63 |
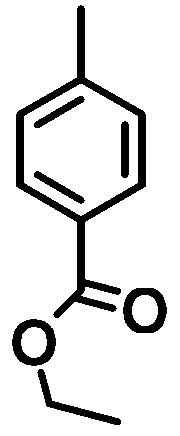
|
410 | 2.42 | Yellow-green | Purple | 178.09 | 94 |
| P64 |

|
420 | 2.12 | Yellow | Black-gray | 212.52 | 94 |

The nitrogen center is the electroactive site of triphenylamine (TPA), linked to three phenyl groups in a propeller-like geometry. This structure means that the A units are incorporated as the side-chains in D–A polymers containing TPA as the D unit. Electron-rich TPA can provide a pair of electrons on the N atom, easily oxidized to form the radical cation at relatively low oxidation potentials.96,97 A significant color difference between the radical cation intermediate during the oxidation process and its corresponding neutral state is observed.98 C. Y. Xu et al.99 designed a series of novel D–A type polymers P65–P67, which contain TPA and ProDOT as D units in the main chain and different electron-withdrawing groups (aldehyde, nitryl, and cyano) as side chains. All the polymer films showed neutral absorption at 400–500 nm with band gaps of about 2.5 eV. The polymer films were yellow or orange in their neutral states and switched to light blue or navy blue in their oxidized states (Fig. 7). In addition, all the polymer films showed strong aggregation-induced fluorescence emission (AIE), seldom reported in the literature. These special characteristics may find applications in fluorescence and fluorescence sensors.
 | ||
| Fig. 7 Chemical structure of P65–P67, spectroelectrochemistry of polymer films on ITO-coated glass substrates in 0.1 M LiClO4/PC at applied potentials, fluorescence photos of P65–P67 in film states under visible light and UV light (λex = 365 nm). Adapted with permission from ref. 99. Copyright 2015 Elsevier. | ||
Z. Q. Liang's recent work100 has also reported a novel D–A polymer P68 (Fig. 8) bearing a TPA donor unit and electron-deficient ketones as a pendant group. This polymer exhibited apparent electrochromism with the color changes under room light from faint yellow to orange, brown, and dark blue. Moreover, it presented simultaneous electrofluorochromic (EFC) behavior with multi-state display properties and remarkably rapid fluorescence from green to the quenched state with a short response time of 0.19 s. Such a rapid response was determined from the ion-diffusion coefficient, which was associated with the nature of the intramolecular charge transfer (ICT) process. In addition, an EFC device based on P68 was successfully assembled, which showed green fluorescence ON/OFF-switching upon applied potentials (in Fig. 8). This work has successfully demonstrated that D–A conjugated polymers bearing TPA as the D unit with appropriate pendant groups can achieve multifunctional performance.
 | ||
| Fig. 8 Chemical structure of P68. (a) UV-vis and color changes under room light (b) PL spectra and fluorescence changes under 365 nm UV light of the P68 film with different applied potentials. (c) Photographs of fluorescence switching in an electrofluorochromic device based on P68. Adapted with permission from ref. 100. Copyright 2016 American Chemical Society. | ||
3.2 Cruciform D–A system
In recent years, donor–acceptor cruciform systems, which are characterized by rigid X-shaped geometry with donors and acceptors on two crossed axes, have been explored to achieve multicolored EC materials. Our group has devoted many efforts to investigate TPA-based D–A cruciform polymers.101,102 All of the three polymers P69–P71 exhibited strong absorption bands centered at about 350 nm, attributed to the π–π* transition of the polymer backbone, exhibiting a neutral yellow color. Upon increasing the voltage gradually, the peak at 350 nm decreased with a shoulder peak at around 490 nm appearing and increasing to a maximum, contributing to the red color. Upon further oxidation, a new strong absorption band at around 760 nm emerged, leading to grey and blue colors. It was intriguing to note that the HOMO is concentrated predominantly on the TPA containing axis, while the LUMO is rearranged along the cyano-containing axis (Fig. 9). The HOMO and LUMO energy levels for such cruciform molecules could be independently tuned through the modulation of structures on the D and A axes. Consequently, the desirable band gap and EC properties can be adjusted. More interestingly, the acceptor axis endows cruciform molecules with both aggregation induced emission effects and high contrast mechanochromic behavior, which shows promising applications for the development of organic optoelectronics such as organic light emitting diodes (OLEDs) and organic photovoltaics (OPVs). | ||
| Fig. 9 (a) Chemical structure of P69–P71, (b) photographs of P69 under natural light and 365 nm UV light, (c) optimized conformations and calculated spatial electron distributions of the HOMO and the LUMO for P69–P71, (d) spectroelectrochemistry and color changes of P69–P71 polymer films. Adapted with permission from ref. 101. Copyright 2014 The Royal Society of Chemistry and ref. 102. Copyright 2015 The Royal Society of Chemistry. | ||
3.3 Dendritic D–A system
Dendritic molecules have attracted much research interest in dye-sensitized solar cells, organic memory devices, and light harvesting materials.103–105 In recent years, there has been an increasing number of research reports on the applications of dendrimers in the EC field.106,107 The particular structure may endow loose aggregation between polymer chains, engendering remarkable EC properties. A. Cihaner and F. Algi et al.108 synthesized two novel D–A compounds P72 and P73 with EDOT and bithiophene as donor units and a central BODIPY core unit as the acceptor part (Fig. 10). Since they have three electroactive polymerizable donor units attached to the central BODIPY structure, which might lead to the formation of a 3D network, facilitating counterion movement into and out of the polymer membranes. It was found that both polymers exhibited low optical band gaps (1.88 eV for P72 and 1.72 eV for P73), resulting in a neutral pink color which switched to blue in the oxidized state. More importantly, P72 displayed a high optical contrast of about 60% in the visible region and a low switching time of 0.5 s. | ||
| Fig. 10 (a) Chemical structures of P72 and P73, (b) schematic representation of the electropolymerization of P72 (c) chronoabsorptometry experiments for P72 on ITO in 0.1 M TBAH/ACN while the polymer was switched between 0.0 and 1.2 V with switch times of 10, 5, and 3 s at 413 nm, 542 nm and 950 nm. Adapted with permission from ref. 108. Copyright 2013 Elsevier. | ||
Among the family of dendritic molecules, dendrimers have well-defined and unique macromolecular structures. Their highly branched arrangements impart important physical and chemical properties which can be different from the linear polymers. The dendrimers containing TPA moieties have attracted wide interest owing to high branching numbers and appear to be more sensitive to electropolymerization than the linear polymers. M. Leung and K. C. Ho et al.109 designed C3-symmetrical dendritic 1,3,5-benzenetricarboxylamides P74. The novel D–A dendrimer is composed of electron-withdrawing carboxyamide groups on the benzene ring as the central core with three electron-donating TPA dendrons as the outer sphere. The structure possesses a combination of hydrogen bonding and aromatic π–π interactions, endowing P74 with combined electroluminescence and electrochromism properties. As shown in Fig. 11, the material exhibited a highly saturated green fluorescence and a colorless film in the neutral state. When the film was oxidized to the first doped state, it turned into brown-red, and meanwhile the photoluminescence was completely quenched in this state. Further oxidation resulted in a change to a deep-blue color. Moreover, the material was highly robust during the redox process.
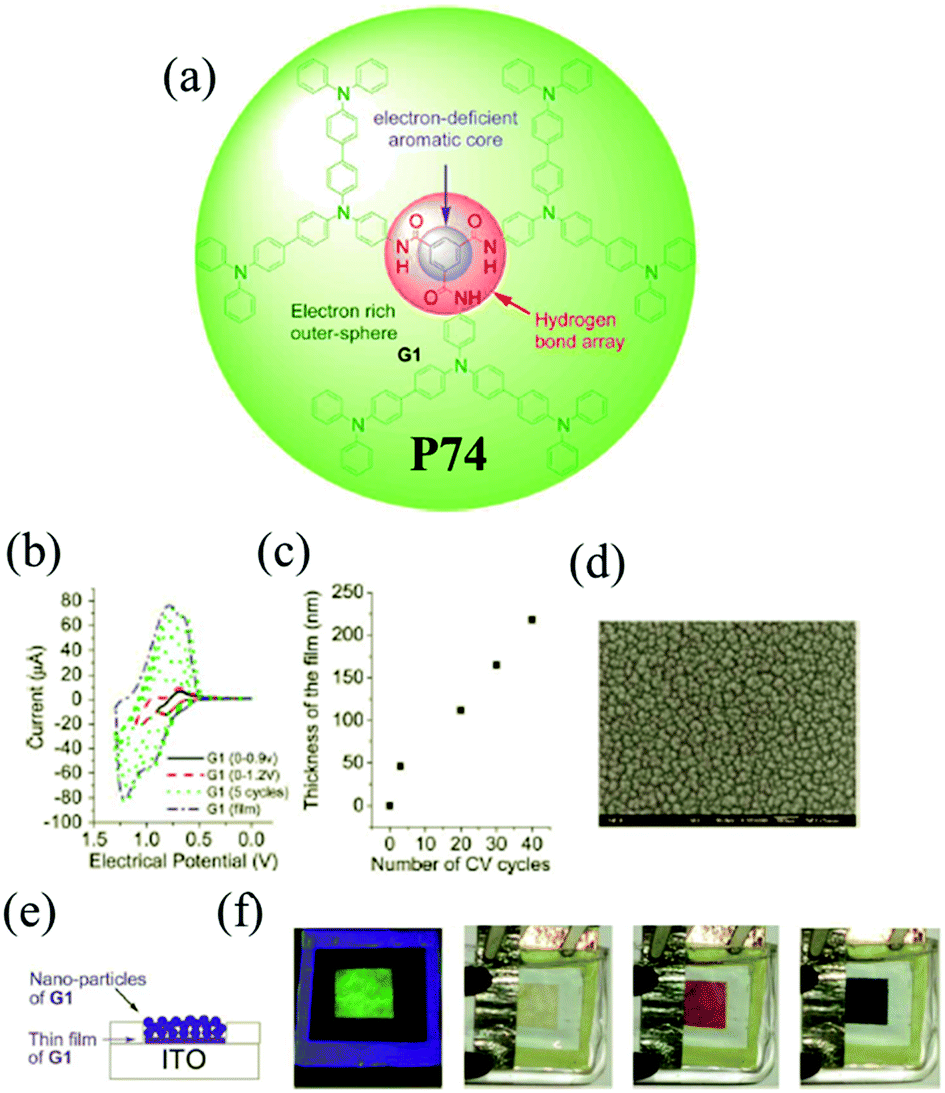 | ||
| Fig. 11 (a) Chemical structure of the monomer of P74, (b) electrochemical characters of the monomer of P74 on Pt in TBAP–CH2Cl2, (c) correlation plot of electropolymerized film thickness and the number of CV cycles, (d) SEM image of electropolymerized P74 on ITO in TBAP–EtCN, (e) schematic diagram of the EC device with nanoparticles of P74 using Bu4NClO4 in CH2Cl2 as supporting electrolyte, (f) green fluorescence and colorless properties of the device at 0 V, brown-red color at 0.9 V, deep blue color at 1.2 V. Adapted with permission from ref. 109. Copyright 2013 The Royal Society of Chemistry. | ||
4. Summary and outlook
In this review, D–A type conjugated polymers with A units in their backbones and side chains, as well as cruciform and dendritic structures have been described in the context of their structural characteristics, color control, EC behaviour and other innovative properties. D–A polymers bearing D and A units in their backbone exhibit tunable band gaps and neutral colors through the introduction of appropriate A units. Their oxidized colors are nonetheless subject to the influence of D units due to the formation of cationic radicals and the doping site for the counterions. Polymers bearing randomly built D units show a combination of the EC characteristics for individual donor systems. The structural characteristics of pyrrole, carbazole and TPA provide the ability to construct D–A polymers with pendent A units. These polymers usually possess relatively high band gaps and thus only small color changes occur, regardless of the A units. Nevertheless, they commonly present a relatively fast response time than most of linear D–A polymers. For innovate D–A systems, including cruciform and dendritic structures, these polymers exhibit tunable colors and excellent EC properties. Other attractive characteristics are also involved in these systems, such as electroluminescence, photoluminescence and mechanochromism. The versatility of cruciform and dendritic D–A polymers may lead to their application in OLEDs, organic field effect transistors and other optoelectronic devices.Despite the achievements in the area of color control through adjusting the species of D or A units and various D–A structures, there are still some fundamental and practical issues that need to be addressed for the next generation of EC polymers. Considering the intrinsic nature of EC polymers, one point is that, the relationship between the conjugated length of the polymer chain and the polaronic character of EC materials (especially the color changes) is still not clear. It is a fundamental problem that is necessary to be solved, because it might allow theoretical predictions and accurate controls of the EC properties of PEC materials in the future. This needs researchers to design and construct a suitable D–A conjugated polymer systems with well-defined conjugation lengths of the polymer chains. Another point we need to pay sufficient attention to is the relationship between the EC switching time and the aggregated state properties in solid for EC materials. A lot of research work has supposed that the fast switching time benefits from the looser packing of the aggregated state for the EC polymer, because it may accelerate the counterion interaction/extraction behaviour, which is a crucial factor for the switching time during the EC processes. But there is still no definite data or quantitative analysis reported to confirm this supposition. Thus designing D–A conjugated microporous polymers with the adjustable aggregated state at the molecular level by introducing the novel porous structure to control the counterion interaction/extraction behaviour, the relationship between the aggregated state and the corresponding switching time may be obtained. The accurate prediction of switching time of EC polymer materials would be expected if it works. In addition, for commercial applications of D–A conjugated polymers, newly emerging film-forming techniques including ink-jet printing and roll-to-roll techniques will be the main trend in the future. Thus the development of soluble polymers via introduction of hydrophilic or hydrophobic groups in the side chain will also be necessary. Meanwhile, the effect of these side-chain functional units on the original EC properties of the conjugated polymers will also be a new issue of concern.
Acknowledgements
The authors gratefully appreciate the suggestions and help from Dr Yaokang Lv, Dr Jingwei Sun and Dr Bin Hu. This work was financially supported by National Natural Science Foundation of China (51673174, 51273179, 51573165, 51403060, 21501148) and Zhejiang Provincial Natural Science Foundation of China (LY15E030006, LQ15E030002).Notes and references
- G. Sonmez, Chem. Commun., 2005, 5251 RSC
.
- G. Sonmez, H. B. Sonmez, C. K. F. Shen and F. Wudl, Adv. Mater., 2004, 16, 1905 CrossRef CAS
- C. L. Gaupp, D. M. Welsh, R. D. Rauh and J. R. Reynolds, Chem. Mater., 2002, 14, 3964 CrossRef CAS
- J. W. Sun, Y. N. Chen and Z. Q. Liang, Adv. Funct. Mater., 2016, 26, 2783 CrossRef CAS
- H. J. Niu, H. Q. Kang, J. W. Cai, C. Wang, X. D. Bai and W. Wang, Polym. Chem., 2011, 2, 2804 RSC
- W. T. Neo, Q. Ye, S. J. Chua and J. W. Xu, J. Mater. Chem. C, 2016, 4, 7364 RSC
- T. Yamase, Chem. Rev., 1998, 98, 307 CrossRef CAS PubMed
- N. M. Rowley and R. J. Mortimer, Sci. Prog., 2002, 85, 243 CrossRef CAS PubMed
- R. J. Mortimer, A. L. Dyer and J. R. Reynolds, Displays, 2006, 27, 2 CrossRef CAS
- A. A. Argun, P. Aubert, B. C. Thompson, I. Schwendeman, C. L. Gaupp, J. Hwang, N. J. Pinto, D. B. Tanner, A. G. MacDiarmid and J. R. Reynolds, Chem. Mater., 2004, 16, 4401 CrossRef CAS
- H. A. M. van Mullekom, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Mater. Sci. Eng., 2001, 32, 1 Search PubMed
- B. C. Thompson, Y. G. Kim, T. D. McCarley and J. R. Reynolds, J. Am. Chem. Soc., 2006, 128, 12714 CrossRef CAS PubMed
- P. J. Shi, C. M. Amb, A. L. Dyer and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2012, 4, 6512 CAS
- C. M. Amb, A. L. Dyer and J. R. Reynolds, Chem. Mater., 2011, 23, 397 CrossRef CAS
- G. Gunbas and L. Toppare, Chem. Commun., 2012, 48, 1083 RSC
- T. Abidin, Q. Zhang, K. L. Wang and D. J. Liaw, Polymer, 2014, 55, 5293 CrossRef CAS
- Z. P. Xu, X. M. Chen, S. Mi, J. M. Zheng and C. Y. Xu, Org. Electron., 2015, 26, 129 CrossRef CAS
- S. Hellström, P. Henriksson, R. Kroon, E. Wang and M. R. Andersson, Org. Electron., 2011, 12, 1406 CrossRef
- E. Oguzhan, H. Bilgili, F. B. Koyuncu, E. Ozdemir and S. Koyuncu, Polymer, 2013, 54, 6283 CrossRef CAS
- A. Balan, D. Baran and L. Toppare, J. Mater. Chem., 2010, 20, 9861 RSC
- G. Q. Ding, C. M. Cho, C. X. Chen, D. Zhou, X. B. Wang, A. Y. X. Tan, J. W. Xu and X. H. Lu, Org. Electron., 2013, 14, 2748 CrossRef CAS
- C. J. Dubois and J. R. Reynolds, Adv. Mater., 2012, 14, 1844 CrossRef
- O. Celikbilek, M. I. Ozkut, F. Algi, A. M. Onal and A. Cihaner, Org. Electron., 2012, 13, 206 CrossRef CAS
- C. M. Cho, Q. Ye, W. T. Neo, T. Lin, X. H. Lu and J. W. Xu, Polym. Chem., 2015, 6, 7570 RSC
- P. J. Shi, C. M. Amb, E. P. Knott, E. J. Thompson, D. Y. Liu, J. G. Mei, A. L. Dyer and J. R. Reynolds, Adv. Mater., 2010, 22, 4949 CrossRef CAS PubMed
- P. Camurlu, T. Durak and L. Toppare, J. Electroanal. Chem., 2011, 661, 359 CrossRef CAS
- G. Hizalan, A. Balan, D. Baran and L. Toppare, J. Mater. Chem., 2011, 21, 1804 RSC
- C. M. Amb, P. M. Beaujuge and J. R. Reynolds, Adv. Mater., 2010, 22, 724 CrossRef CAS PubMed
- S. Chen, D. Zhang, M. Wang, L. Q. Kong and J. S. Zhao, New J. Chem., 2016, 40, 2178 RSC
- S. Das, P. B. Pati and S. S. Zade, Macromolecules, 2012, 45, 5410 CrossRef CAS
- C. X. Xua, J. S. Zhao, M. Wang, Z. Wang, C. S. Cui, Y. Kong and X. X. Zhang, Electrochim. Acta, 2012, 75, 28 CrossRef
- A. C. Ozelcaglayan, M. Sendur, N. Akbasoglu, D. Hazar Apaydin, A. Cirpan and L. Toppare, Electrochim. Acta, 2012, 67, 224 CrossRef CAS
- D. Baran, A. Balan, S. Celebi, B. M. Esteban, H. Neugebauer, N. S. Sariciftci and L. Toppare, Chem. Mater., 2010, 22, 2978 CrossRef CAS
- Q. Ye, W. T. Neo, T. Lin, J. Song, H. Yan, H. Zhou, K. W. Shah, S. J. Chua and J. W. Xu, Polym. Chem., 2015, 6, 1487 RSC
- Q. Ye, W. T. Neo, C. M. Cho, S. W. Yang, T. T. Lin, H. Zhou, H. Yan, X. H. Lu, C. Y. Chi and J. W. Xu, Org. Lett., 2014, 16, 6386 CrossRef CAS PubMed
- E. Sefer, H. Bilgili, B. Gultekin, M. Tonga and S. Koyuncu, Dyes Pigm., 2015, 113, 121 CrossRef CAS
- S. Tarkuc, Y. A. Udum and L. Toppare, Polymer, 2009, 50, 3458 CrossRef CAS
- C. B. Fan, C. Q. Ye, X. M. Wang, Z. G. Chen, Y. Y. Zhou, Z. Q. Liang and X. T. Tao, Macromolecules, 2015, 48, 6465 CrossRef CAS
- M. A. Anber, B. Milde, W. Alhalasah, H. Lang and R. Holze, Electrochim. Acta, 2008, 53, 6038 CrossRef
- E. Varathan, D. Vijay and V. Subramanian, J. Phys. Chem. C, 2014, 118, 21741 CAS
- S. S. Zhang, Z. X. Qu, P. Tao, B. Brooks, Y. H. Shao, X. Y. Chen and C. G. Liu, J. Phys. Chem. C, 2012, 116, 12434 CAS
- S. M. Link, M. Scheuble, M. Goll, E. Muks, A. Ruff, A. Hoffmann, T. V. Richter, J. T. L. Navarrete, M. C. R. Delgado and S. Ludwigs, Langmuir, 2013, 29, 15463 CrossRef CAS PubMed
- B. C. Wanga, H. R. Liao, J. C. Chang, L. Chen and J. T. Yeh, J. Lumin., 2007, 124, 333 CrossRef
- A. Semaözen, C. Atilgan and G. Sonmez, J. Phys. Chem. C, 2007, 111, 16362 Search PubMed
- Y. Furukawa, J. Phys. Chem., 1996, 100, 15644 CrossRef CAS
- O. Atwani, C. Baristiran, A. Erden and G. Sonmez, Synth. Met., 2008, 158, 83 CrossRef CAS
- M. I. Ozkut, M. P. Algi, Z. Oztas, F. Algi, A. M. Önal and A. Cihaner, Macromolecules, 2012, 45, 729 CrossRef CAS
- Z. Xu, M. Wang, J. S. Zhao, C. S. Cui, W. Y. Fan and J. F. Liu, Electrochim. Acta, 2014, 125, 241 CrossRef CAS
- B. Yigitsoy, S. M. A. Karim, A. Balan, D. Baran and L. Toppare, Electrochim. Acta, 2011, 56, 2263 CrossRef CAS
- E. Sefer and F. B. Koyuncu, Electrochim. Acta, 2014, 143, 106 CrossRef CAS
- E. Rustamlı, S. Goker, S. Tarkuc, Y. A. Udum and L. Toppare, J. Electrochem. Soc., 2015, 162, G75 CrossRef
- E. K. Unver, S. Tarkuc, Y. A. Udum, C. Tanyeli and L. Toppare, Org. Electron., 2011, 12, 1625 CrossRef CAS
- C. Kitamura, S. Tanaka and Y. Yamashita, Chem. Mater., 1996, 8, 570 CrossRef CAS
- G. Sonmez, C. K. F. Shen, Y. Rubin and F. Wudl, Angew. Chem., Int. Ed., 2004, 43, 1498 CrossRef CAS PubMed
- S. Ozdemir, M. Sendur, G. Oktem, O. Dogan and L. Toppare, J. Mater. Chem., 2012, 22, 4687 RSC
- H. Zhao, Y. Y. Wei, J. S. Zhao and M. Wang, Electrochim. Acta, 2014, 146, 231 CrossRef CAS
- L. B. Greoenendaal, G. Zptti, P. H. Aubert, S. M. Waybright and J. R. Reynold, Adv. Mater., 2013, 15, 855 CrossRef
- A. Kumar, D. M. Welsh, M. C. Morvant, F. Piroux, K. A. Abboud and J. R. Reynolds, Chem. Mater., 1998, 10, 896 CrossRef CAS
- B. C. Thompson, P. Schottland, K. Zong and J. R. Reynolds, Chem. Mater., 2000, 12, 1563 CrossRef CAS
- V. K. Thakur, G. Q. Ding, J. Ma, P. S. Lee and X. H. Lu, Adv. Mater., 2012, 24, 4071 CrossRef CAS PubMed
- A. Balan, G. Gunbas, A. Durmus and L. Toppare, Chem. Mater., 2008, 20, 7510 CrossRef CAS
- H. Akpınar, A. Balan, D. Baran, E. K. Ünver and L. Toppare, Polymer, 2010, 51, 6123 CrossRef
- D. J. Irvin, C. J. DuBois and J. R. Reynolds, Chem. Commun., 1999, 2121 RSC
- C. J. Dubois, K. A. Abboud and J. R. Reynolds, J. Phys. Chem. B, 2004, 108, 8550 CrossRef CAS
- G. Sonmez, H. Meng and F. Wudl, Chem. Mater., 2004, 16, 574 CrossRef CAS
- P. Camurlu, T. Durak, A. Balan and L. Toppare, Synth. Met., 2011, 161, 1898 CrossRef CAS
- A. Durmus, G. E. Gunbas and L. Toppare, Chem. Mater., 2007, 19, 6247 CrossRef CAS
- G. E. Gunbas, A. Durmus and L. Toppare, Adv. Mater., 2008, 20, 691 CrossRef CAS
- G. E. Gunbas, A. Durmus and L. Toppare, Adv. Funct. Mater., 2008, 18, 2026 CrossRef CAS
- F. Algı and A. Cihaner, Org. Electron., 2009, 10, 704 CrossRef
- A. Durmus, G. E. Gunbas, P. Camurlu and L. Toppare, Chem. Commun., 2007, 3246 RSC
- D. M. Welsh, A. Kumar, E. W. Meijer and J. R. Reynolds, Adv. Mater., 1999, 11, 1379 CrossRef CAS
- C. L. Gaupp, D. M. Welsh and J. R. Reynolds, Macromol. Rapid Commun., 2002, 23, 885 CrossRef CAS
- D. M. Welsh, L. J. Kloeppner, L. Madrigal, M. R. Pinto, B. C. Thompson, K. S. Schanze, K. A. Abboud, D. Powell and J. R. Reynolds, Macromolecules, 2002, 35, 6517 CrossRef CAS
- M. Icli, M. Pamuk, F. Algi, A. M. Önal and A. Cihaner, Chem. Mater., 2010, 22, 4034 CrossRef CAS
- M. I. Ozkut, M. P. Algi, Z. Oztas, F. Algi, A. M. Önal and A. Cihaner, Macromolecules, 2012, 45, 729 CrossRef CAS
- S. Varis, M. Ak, C. Tanyeli, I. M. Akhmedov and L. Toppare, Eur. Polym. J., 2006, 42, 2352 CrossRef CAS
- E. Yildiz, P. Camurlu, C. Tanyeli, I. Akhmedov and L. Toppare, J. Electroanal. Chem., 2008, 612, 247 CrossRef CAS
- S. Tarkuc, E. Sahmetlioglu, C. Tanyeli, I. M. Akhmedov and L. Toppare, Electrochim. Acta, 2006, 51, 5412 CrossRef CAS
- S. Celebi, A. Balan, B. Epik, D. Baran and L. Toppare, Org. Electron., 2009, 10, 631 CrossRef CAS
- A. T. Taskin, A. Balan, B. Epik, E. Yildiz, Y. A. Udum and L. Toppare, Electrochim. Acta, 2009, 54, 5449 CrossRef CAS
- D. Baran, G. Oktem, S. Celebi and L. Toppare, Macromol. Chem. Phys., 2011, 212, 799 CrossRef CAS
- D. Witker and J. R. Reynolds, Macromolecules, 2005, 38, 7636 CrossRef CAS
- Y. A. Udum, C. G. Hızlıateş, Y. Ergün and L. Toppare, Thin Solid Films, 2015, 595, 61 CrossRef CAS
- F. B. Koyuncu, Electrochim. Acta, 2012, 68, 184 CrossRef CAS
- C. X. Xu, J. S. Zhao, M. Wang, C. S. Cui and R. M. Liu, Thin Solid Films, 2013, 527, 232 CrossRef CAS
- P. M. Beaujuge, S. Ellinger and J. R. Reynolds, Nat. Mater., 2008, 7, 795–799 CrossRef CAS PubMed
- P. M. Beaujuge, S. V. Vasilyeva, D. Y. Liu, S. Ellinger, T. D. McCarley and J. R. Reynolds, Chem. Mater., 2012, 24, 255–268 CrossRef CAS
- W. T. Neo, L. M. Loo, J. Song, X. B. Wang, C. M. Cho, H. S. O. Chan, Y. Zong and J. W. Xu, Polym. Chem., 2013, 4, 4663 RSC
- B. He, W. T. Neo, T. L. Chen, L. M. Klivansky, H. X. Wang, T. W. Tan, S. J. Teat, J. W. Xu and Y. Liu, ACS Sustainable Chem. Eng., 2016, 4, 2797 CrossRef CAS
- M. Sendur, A. Balan, D. Baran, B. Karabay and L. Toppare, Org. Electron., 2010, 11, 1877 CrossRef CAS
- J. Hwang, J. I. Son and Y. B. Shim, Sol. Energy Mater. Sol. Cells, 2010, 94, 1286 CrossRef CAS
- A. Cihaner and F. Algi, Electrochim. Acta, 2008, 54, 786 CrossRef CAS
- B. Hu, X. J. Lv, J. W. Sun, G. F. Bian, M. Ouyang, Z. Y. Fu, P. J. Wang and C. Zhang, Org. Electron., 2013, 14, 1521 CrossRef CAS
- B. Hu, Y. J. Zhang, X. J. Lv, M. Ouyang, Z. Y. Fu and C. Zhang, J. Electroanal. Chem., 2013, 689, 291 CrossRef CAS
- S. Beaupré, J. Dumas and M. Leclerc, Chem. Mater., 2006, 18, 4011 CrossRef
- L. L. Ji, Y. Y. Dai, S. M. Yan, X. J. Lv, C. Su, L. H. Xu, Y. K. Lv, M. Ouyang, Z. F. Chen and C. Zhang, Sci. Rep., 2016, 6, 30068 CrossRef PubMed
- M. H. Tremblay, T. Skalski, Y. Gautier, G. Pianezzola and W. G. Skene, J. Phys. Chem. C, 2016, 120, 9081 CAS
- S. Mi, J. C. Wu, J. Liu, J. M. Zheng and C. Y. Xu, Org. Electron., 2015, 23, 116 CrossRef CAS
- J. W. Sun and Z. Q. Liang, ACS Appl. Mater. Interfaces, 2016, 8, 18301 CAS
- J. W. Sun, X. J. Lv, P. J. Wang, Y. J. Zhang, Y. Y. Dai, Q. C. Wu, M. Ouyang and C. Zhang, J. Mater. Chem. C, 2014, 2, 5365 RSC
- J. W. Sun, Y. Y. Dai, M. Ouyang, Y. J. Zhang, L. L. Zhan and C. Zhang, J. Mater. Chem. C, 2015, 3, 3356 RSC
- R. J. Dong, Y. F. Zhou and X. Y. Zhu, Acc. Chem. Res., 2014, 47, 2006 CrossRef CAS PubMed
- X. Yang, H. Shang, C. Ding and J. Li, Polym. Chem., 2015, 6, 668 RSC
- Q. Wei, K. Achazi, H. Liebe, A. Schulz, P. L. M. Noeske, I. Grunwald and R. Haag, Angew. Chem., Int. Ed., 2014, 53, 11650 CrossRef CAS PubMed
- M. Sezgin, O. Ozay, S. Koyuncu, H. Ozay and F. B. Koyuncu, Chem. Eng. J., 2015, 274, 282 CrossRef CAS
- S. Y. Kao, Y. S. Lin, C. W. Hu, M. K. Leung and K. C. Ho, Sol. Energy Mater. Sol. Cells, 2015, 143, 174 CrossRef CAS
- M. P. Algia, S. Tirkesb, S. Ertanb, E. G. C. Ergunb, A. Cihaner and F. Algi, Electrochim. Acta, 2013, 109, 766 CrossRef
- M. K. Leung, Y. S. Lin, C. C. Lee, C. C. Chang, Y. X. Wang, C. P. Kuo, N. Singh, K. R. Lin, C. W. Hu, C. Y. Tseng and K. C. Ho, RSC Adv., 2013, 3, 22219 RSC
| This journal is © The Royal Society of Chemistry 2017 |