
Photocontrolled protein assembly for constructing programmed two-dimensional nanomaterials†
Linlu
Zhao
,
Yijia
Li
,
Tingting
Wang
,
Shanpeng
Qiao
,
Xiumei
Li
,
Ruidi
Wang
,
Quan
Luo
 ,
Chunxi
Hou
,
Jiayun
Xu
and
Junqiu
Liu
*
,
Chunxi
Hou
,
Jiayun
Xu
and
Junqiu
Liu
*
State Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, China. E-mail: junqiuliu@jlu.edu.cn
First published on 28th November 2017
Abstract
Precise self-assembly of proteins with structural heterogeneity, flexibility, and complexity into programmed arrays to mimic the exquisite architectures created by Nature is a great challenge for the development of protein-based functional nanomaterials. Herein, we present a strategy that integrates light stimuli and covalent coupling to prepare size-tunable two-dimensional (2D) protein nanostructures by remote photocontrol. Using Ru(bpy)32+ as a photosensitizer, stable protein one (SP1) was redesigned and self-assembled into nanosheets in the presence of ammonium persulfate (APS) through a rapid and efficient oxidative protein crosslinking reaction. In the design, only a serine-to-tyrosine mutation at position 98 was introduced into SP1 by combining computer simulation and genetic engineering for specific covalent coupling under white light illumination. The chemical and topographical specificities of the photosensitized crosslinking reaction allow control of the direction of protein assembly to form extended 2D nanosheets, which are packed in an orderly manner along the lateral surface of ring-shaped SP1S98Y. Notably, the growth of SP1 nanosheets exhibited isotropical characteristics and can be dynamically mediated by illumination time to achieve precise control of the size of the assembled architectures. The subsequent heat treatment further revealed the excellent thermostability of the 2D periodic SP1 nanostructures, which may find promising applications in the fabrication of various nanobiomaterials after functionalization. The present work demonstrates that the visible light-triggered crosslinking strategy is a facile and environmentally friendly method for constructing advanced protein architectures through hierarchical self-assembly.
Introduction
Most proteins in cells perform their biological functions in the form of large complexes with diverse extended structures such as fibers, sheets, and multidimensional nano-/macro-arrays.1 These large-scale protein systems often exhibit unique advantages in terms of physical/chemical properties, including stability for structural support, dynamic assembly/disassembly for intelligent transformation, special microenvironments for enzymatic catalysis, etc.2,3 Following the wisdom of nature, the construction of various hierarchical protein nanostructures in vitro has become a significant area of research over the past decades for contemporary researchers in understanding and developing novel nanostructures and nanobiomaterials.4–11 An interesting question is how to control individual proteins to create the desired modular and hierarchical architectures.In the field of protein assembly, there mainly exist two fascinating tendencies: one is establishing novel strategies to induce protein assembly into highly ordered architectures,12–17 where various interactions such as electrostatic interactions,18–21 metal–ligand coordination,22–24 host–guest interactions,25–27 and chemical cross-linking28 have been widely adopted to fabricate complex superstructures including nanowires,29,30 nanosheets,31,32 nanorings,33 and network structures.34,35 The other is utilizing the response to various external stimuli, such as light, temperature, and pH, to realize regulation in the protein assembly/disassembly process or reversible morphology interconversion.36–38 However, the integration of the above two aspects in fabricating hierarchical protein nanobiomaterials has rarely been reported, as few proteins can achieve assembly and regulation simultaneously under specified conditions. Photocatalyst-mediated protein self-assembly may provide a new strategy for realizing the controllable growth of protein nanostructures.
Stable protein one (SP1), isolated from aspen plants (Populus tremula), is a cricoid protein consisting of twelve subunits bound to each other via hydrophobic interactions to afford a double-layered nanoring.39,40 From the crystal structure of SP1 we know that the outer diameter, inner diameter, and width are about 11 nm, 2.5 nm, and 4.5 nm, respectively (Scheme 1a). Notably, the specific C6 symmetric structure endows SP1 with great potential in constructing hierarchical architectures under rational design. In our previous work, we have successfully demonstrated that enzyme-triggered nanotechnology can direct the assembly behavior with SP1 as a building block model. As horse radish peroxidase (HRP) was known to catalyze the substrate tyrosine residues (Tyr) with high specificity, it was feasible to utilize the HRP-induced covalent protein assembly of cricoid SP1 to form nanosheets. As predicted, the Tyr–Tyr coupling made it possible for multiple SP1 rings to interact with each other isotropically at the lateral surface, further controlling the assembly orientation into two-dimensional sheet-like protein nanoarrays.41
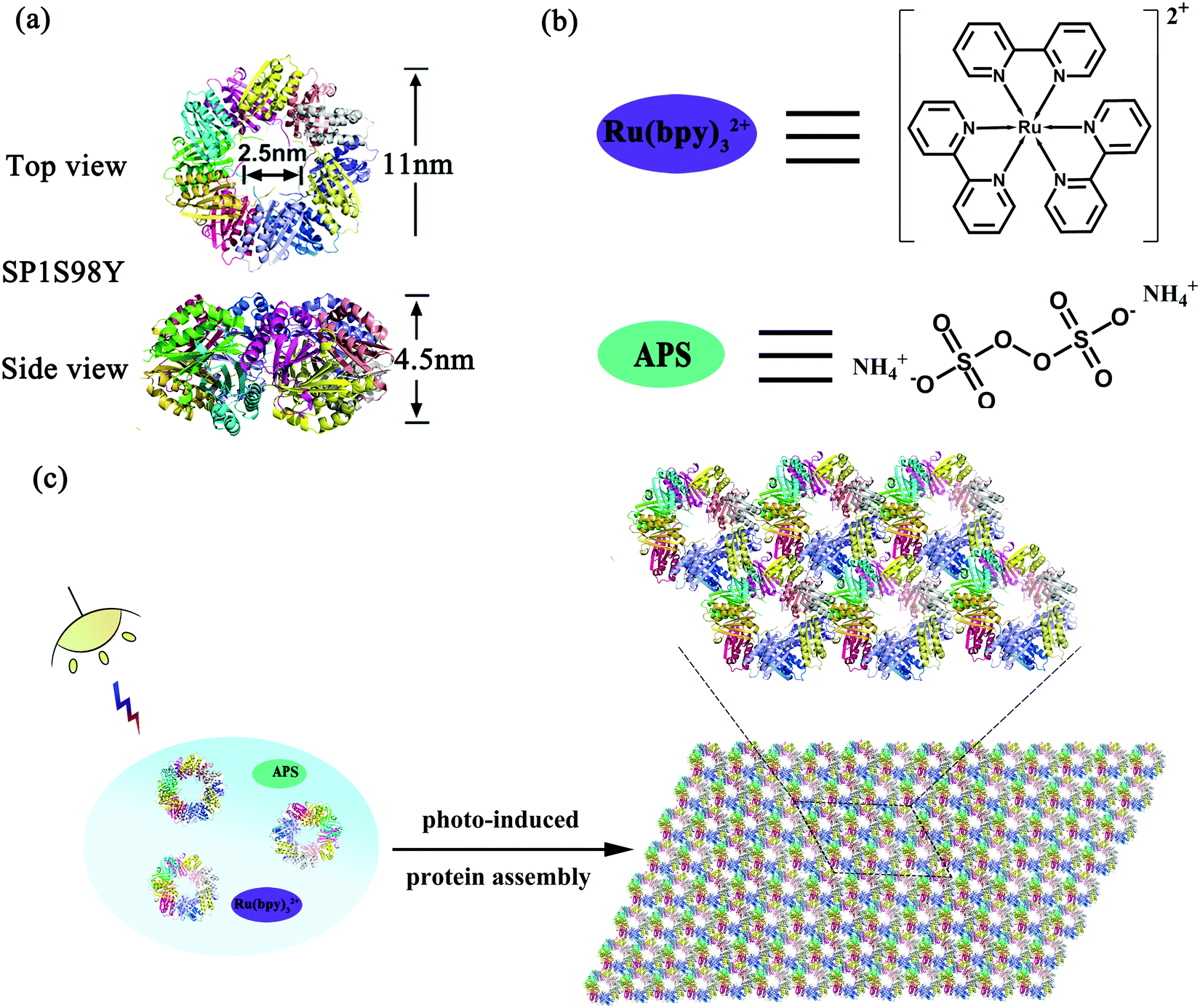 | ||
| Scheme 1 The schematic representation of photo-induced protein assembly. (a) The top and side views of the SP1S98Y nanoring; (b) the structure of Ru(bpy)32+ and APS; (c) coassembly of SP1S98Y, Ru(bpy)32+ and APS to construct 2D nanosheets upon illumination. | ||
The successful research validated the feasibility of covalent protein assembly into highly ordered nanostructures. However, it underwent free assembly procedures once the enzyme-catalyzed process started, that is to say, the size of the assemblies cannot be controlled in this system, which may restrict its practical applications. We are thus curious about whether external stimuli can be introduced in the protein assembly system to serve as an efficient tool for precisely controlling the size of the assemblies. Among various environmental stimuli, light is considered to be the optimum candidate not only for its excellent performance in triggering the growth of green plants, but also for its noninvasive controlling properties. Inspired by the wonderful wisdom of nature, herein we developed a novel strategy for the construction of photocontrolled 2D protein nanosheets with precisely regulated morphology through a photocatalyst-induced protein assembly. Ruthenium(II) tris-bipyridyl dication (Ru(bpy)32+), affording efficient catalytic coupling of specific subunits under light stimuli, may be the best and simplest photocatalyst to evoke the assembly process (Scheme 1b).42,43 For the protein system, Ru(bpy)32+ possessed the ability to catalyze two specific amino acids, cysteine (Cys) and tyrosine (Tyr) residues, under mild visible light. Notably, the addition of ammonium persulfate (APS) can largely improve the photocatalytic efficiency (Scheme 1b). Fortunately, wild-type SP1 had no Cys residues among the whole sequence and several Tyr residues incorporated inside with rather low reactive activity, which favored the artificial design of this charming protein model. According to these prominent features, we anticipated that the modified protein SP1 with Tyr residues introduced to the target site could guide the assembly orientation to eventually form orderly arranged sheet nanoarrays upon Ru(bpy)32+ catalysis (Scheme 1c). The constructed versatile assembly scaffolds held the potential to be optically regulated as illumination was considered as the main prerequisite for the occurrence of protein–protein ordered coupling and also played a pivotal role in controlling this assembly process. Therefore, we further developed light-stimulated growth of the assemblies, achieving precise control over the nanostructures in the field of protein assembly.
Result and discussion
In order to achieve efficient protein assembly by Ru(bpy)32+ catalysis, we first endeavored to rationally design the model protein SP1. With the aid of computer simulation and genetic engineering, the Ser98 at the lateral surface of ring-shaped SP1 was selected as an active site to introduce Tyr residues, which was believed to be the most suitable position for effective covalent coupling. Significantly, when the cricoid SP1S98Y proteins moved close to each other via the interactions among the above site, it can realize charge complementation between the adjacent protein to a large extent, and thus facilitated the ordered assembly into desired architectures (Fig. S1, ESI†). The purification and identification of the resulting SP1 variant were performed by SDS-PAGE (Fig. S2, ESI†) and MALDI-TOF mass spectrometry (Fig. S3, ESI†), respectively. Also, circular dichroism (CD) data showed no significant transition of the secondary structure of the SP1 mutant compared to wild-type SP1 (Fig. S4, ESI†).The original Tyr residues inside SP1S98Y may participate in covalent coupling under high concentration of photocatalyst Ru(bpy)32+, which would disturb the assembly orientation. Therefore, the primary task is to investigate the optimum catalytic conditions for ensuring the assembly process as designed. From the above analyses, we should keep in mind that the essential requirement was holding the concentration of Ru(bpy)32+ to both guarantee the effective coupling and avoid the undesired reactive activity. We then carried out a series of assembly attempts with the concentration of Ru(bpy)32+ ranging from 10−6 to 10−4 M, and APS from 2.5 × 10−5 to 2.5 × 10−3 M respectively under white light illumination for 20 min. The covalent assembly behaviors were first investigated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis (Fig. 1a). The obtained SP1 dimers were attributed to the intermolecular covalent coupling among adjacent SP1S98Y proteins, which was supposed to be Tyr–Tyr coupling of the introduced target site. However, when increasing the photocatalyst to a relatively high concentration, the assembly system can even find multimers from SDS-PAGE, which indicated the participation of the original Tyr residues inside SP1S98Y to achieve random coupling, thus harassing the assembly process. Therefore, in order to enable the oriented assembly, we fixed the concentrations of Ru(bpy)32+ and APS to be 5 × 10−6 M and 1.25 × 10−4 M respectively for the following photo-induced protein assembly system.
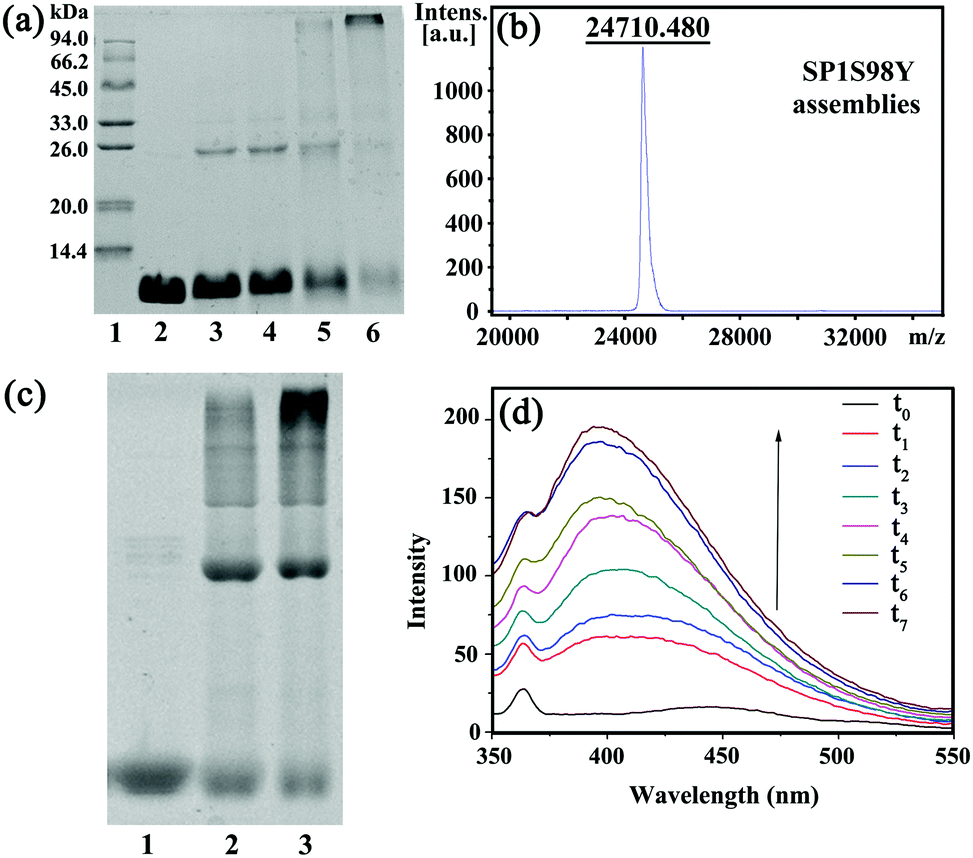 | ||
| Fig. 1 (a) 15% SDS-PAGE analysis of the SP1S98Y-based photo-induced assembly. Lane 1: protein marker; lane 2: purified SP1S98Y; lanes 3 to 6 refer to the crosslinking assemblies with various molar concentrations of Ru(bpy)32+ (10−6 M, 5 × 10−6 M, 10−5 M, 10−4 M, respectively) and APS (2.5 × 10−5 M, 1.25 × 10−4 M, 2.5 × 10−4 M, 2.5 × 10−3 M, respectively); (b) MALDI-TOF mass spectrometry analysis of the assemblies; (c) 8% native-PAGE analysis of free SP1S98Y (lane 1) and photo-induced assemblies with an illumination time of 15 min (lane 2) and 30 min (lane 3); (d) fluorescence emission spectra of SPS98Y-based assemblies (λex = 325 nm). t0–t7 respectively are SP1S98Y + Ru(bpy)32+ + APS with white light illumination for 0 min, 10 min, 20 min, 30 min, 40 min, 50 min, 60 min, and 70 min. [SP1S98Y] = 10−5 M (cal. by monomer). | ||
MALDI-TOF mass spectrometry was also utilized to characterize the assemblies. The obtained molecular weight consistent with the SP1S98Y dimer (Fig. 1b) was strong proof of the covalent cross-linking between the proteins compared to the only monomer observed in SP1S98Y as a control (Fig. S3, ESI†). Furthermore, the native-PAGE analysis demonstrated the assembly behaviors of the SP1S98Y-based photocatalytic system. As shown in Fig. 1c, the assembled samples exhibited some new bands at high molecular weights in comparison with the free SP1S98Y, which was indicative of effective crosslinking. Fortunately, the generated Tyr-coupled product possessed the fluorescence characteristics of the assembled dimer.44,45 As expected, there appeared a characteristic peak of 405 nm in the fluorescence emission spectra upon the addition of Ru(bpy)32+ and APS to the assembly system and then illumination with white light. As time went on, the intensity of the peak increased and eventually became essentially constant after illumination for 1 h, which illustrated the process of effective Tyr–Tyr coupling under a light stimulus (Fig. 1d).
Dynamic light scattering (DLS) was subsequently utilized to investigate the process of protein self-assembly and understand the significant roles of the photocatalyst and light. From the typical number-averaged DLS curves shown in Fig. 2a, the hydrodynamic diameter of the SP1S98Y protein alone showed only one peak at about 10 nm, which was in good agreement with its crystal size. In contrast, after incubation with appropriate concentrations of Ru(bpy)32+ and APS for different illumination times, the peak consistent with the free SP1S98Y completely disappeared and different assembly peaks with increasing sizes were detected subsequently, indicating the formation of SP1S98Y assemblies. To further explore the growing process of protein assembly, kinetic curves were obtained according to the time-dependent DLS analysis, which enabled us to have a better understanding of the significance of the illumination time to the assembly sizes (Fig. 2b). Another study was carried out to investigate the influence of reaction conditions on the occurrence of assembly (Fig. S5, ESI†). In the control group, when the coassembled system was absent of either light illumination or photocatalyst Ru(bpy)32+, the size of the assemblies in the DLS curves seemed almost unchanged compared to the pure SP1S98Y even after rather a long time, demonstrating the significant roles of these two factors in controlling the assembly process. Interestingly, in the absence of APS, the assembly size increased but quite a bit slower than in the normal group within the same time. Furthermore, we also used wild-type SP1 as a model protein to conduct assembly as with SP1S98Y. The phenomenon of no obvious change in size in the control system revealed that the SP1S98Y-based system was believed to achieve assembly due to the introduced target Tyr–Tyr coupling.
 | ||
| Fig. 2 (a) The dynamic light scattering (DLS) analysis of the hydrodynamic diameters of the SP1S98Y-based assembly system with white light illumination for 0 min (orange), 5 min (black), 10 min (blue), 15 min (dark yellow), 20 min (purple), 30 min (olive), 40 min (green), 50 min (pink), 60 min (red), and 70 min (green); (b) kinetic curve of the formation of protein assemblies obtained from DLS studies. | ||
After the DLS analysis had preliminarily validated the size-increasing tendency upon photo-catalyzing protein assembly, the morphology of the corresponding architectures was then explored by tapping-mode atomic force microscopy (AFM). As shown in Fig. 3a, when the coassembled system was illuminated with white light for 15 min, the original random dispersed SP1S98Y nanoring with Tyr residues at the periphery started to grow in lateral directions to form 2D nanosheets of about 150 nm in size. The uniform height (4.3 nm) of the obtained nanostructures matched well with the theoretical size of SP1 in the crystal structure (width 4.5 nm), indicating the successful construction of single-layer architectures (Fig. 3b). Also, the 3D image of the sheet structures gave further support for this definite morphology (Fig. 3c). Upon extending light illumination for 30 min, the cricoid protein again generated highly ordered 2D layer-like nanostructures but possessed a larger assembled size of approximately 500 nm (Fig. 3d). Similarly, the obtained nanosheets exhibited identical height (Fig. 3e) and uniformity (Fig. 3f) as before, illustrating the continuous growth of the protein superstructures along the lateral side of the ring-shaped SP1S98Y under light stimuli. The data above clearly identified that the obtained nanosheets were consistent well with our original design and at the same time confirmed the feasibility of photo-induced nanotechnology in catalyzing protein systems to achieve superstructures.
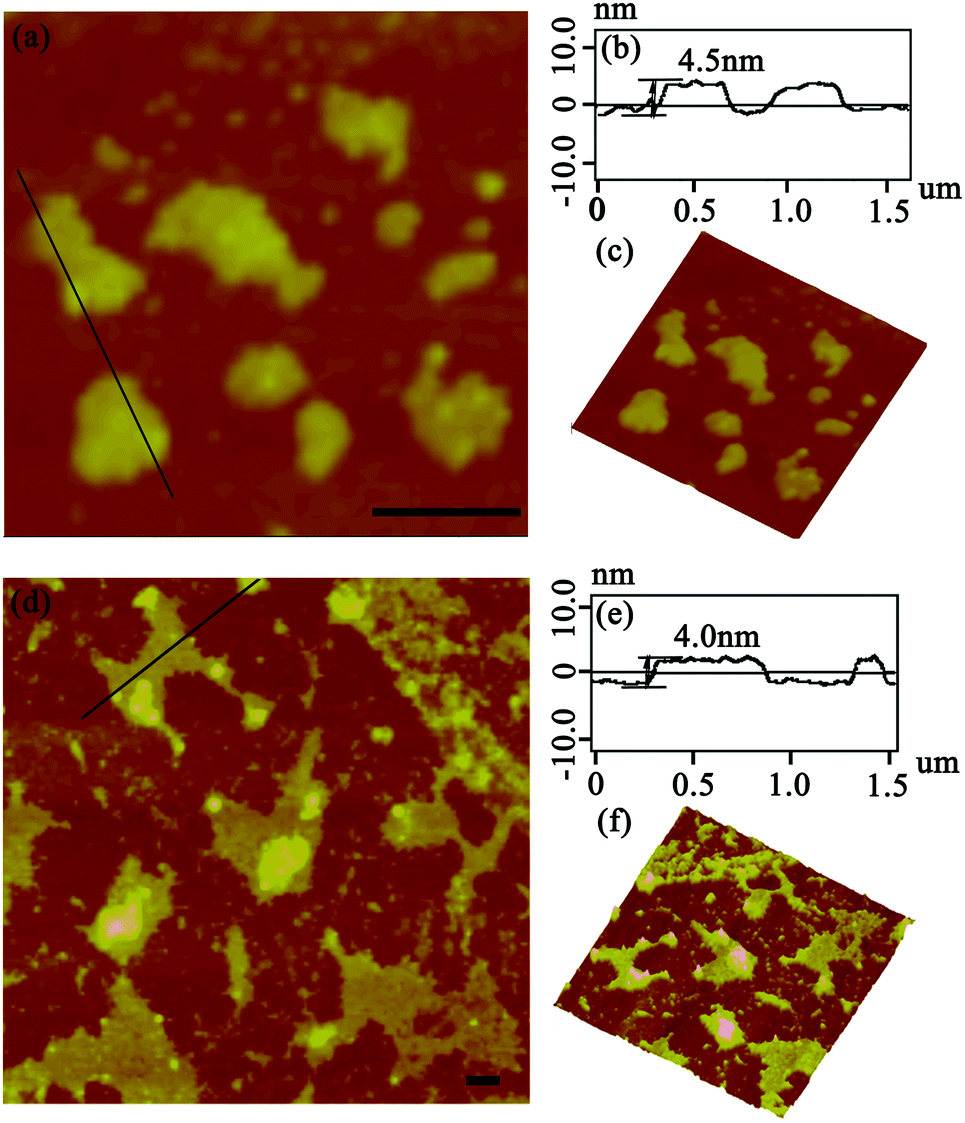 | ||
| Fig. 3 Morphologies of SP1S98Y-based nanostructures. (a and d) AFM images of the assembled 2D nanosheets illuminated for 15 min and 30 min, respectively; (b and e) associated height profile along the black line in panels (a) and (d); (c and f) 3D images of the structures in (a) and (d). Both the scale bars represent 200 nm. | ||
As this protein assembly process was triggered by light stimuli, we thus had a strong desire to investigate in detail the effect of illumination time on the morphology, especially for size control. The AFM images gave us further favorable evidence for the photo-controlled protein self-assembly behavior. When the illumination time was restricted to 45 min, the assembly system also exhibited sheet-like nanostructures of a highly ordered arrangement pattern. The size of the assemblies can be controlled to nearly 800 nm in comparison with the preceding architectures (Fig. 4a). Notably, the assembly process always grew isotropically along the periphery of cricoid SP1S98Y upon light stimuli to achieve single-layered nanoarrays, highlighting the guidance of the introduced Tyr residues for obtaining the desired superstructures in our design (Fig. 4b and c). The initial characterizations revealed that the light-induced assembly tended to complete within 60 min, which was assumed to be restricted by the final concentration of the model protein and APS. The ultimate assembly morphology was also investigated by AFM. Surprisingly, the assemblies possessed the ability to form prominent well-distributed 2D nanosheets of micrometer size and at the same time uniform height close to the crystal structure (Fig. 4d–f). These studies well proved the successful realization of programmed assembly procedures in controlling the architectures for designed sheet-like nanostructures.
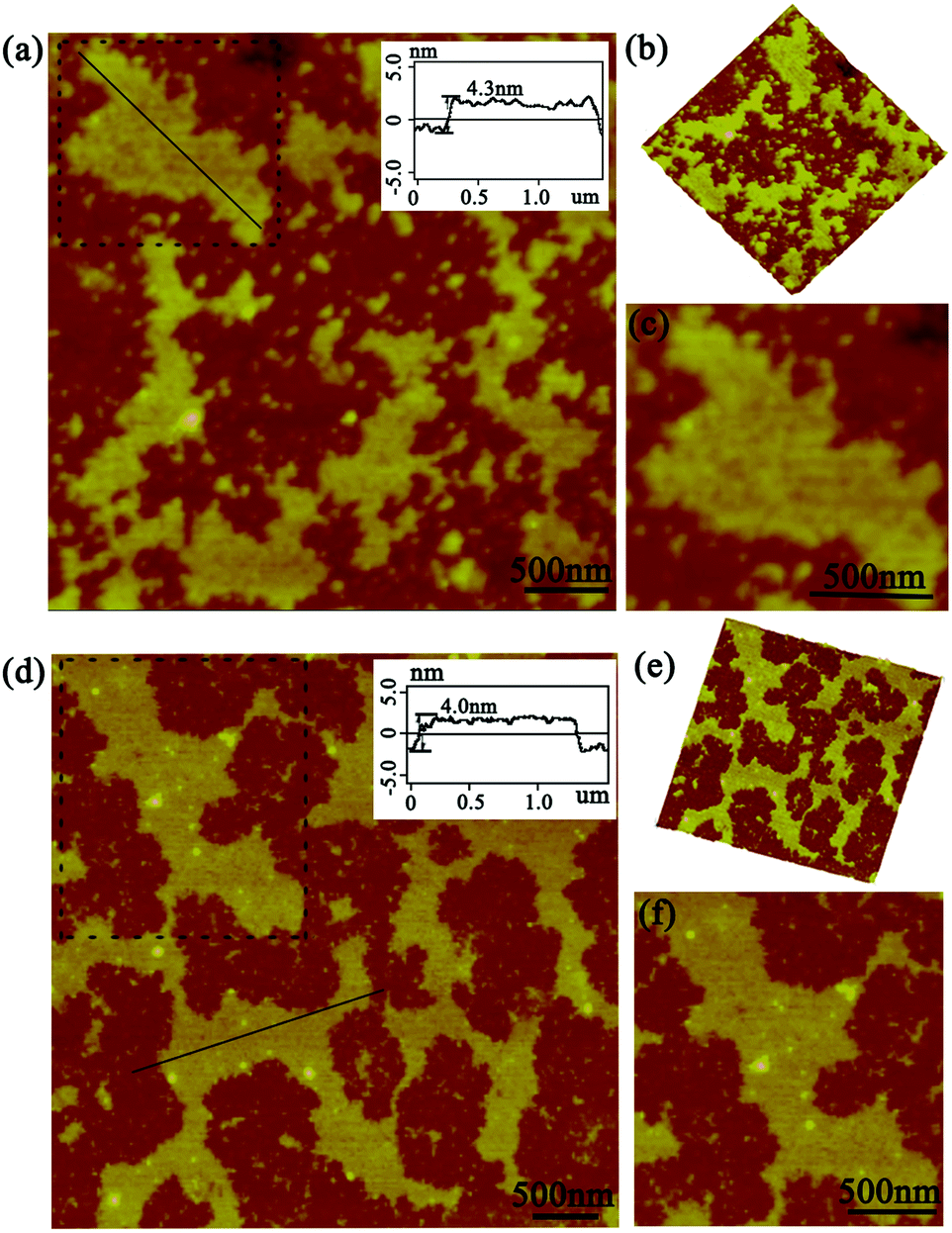 | ||
| Fig. 4 AFM images of the photo-induced assemblies. (a and d) The topographical images with illumination times of 45 min and 60 min, respectively; (b and e) 3D images of the structures in (a and d); (c and f) the detailed structures of the squares in (a) and (d); the inset showed the associated height profile along the black line in panels (a and d). | ||
To further explore the detailed structure of the assemblies, scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were carried out. The SEM images well supported the AFM findings. As illumination time prolonged, the generated 2D nanosheets grew gradually from the early 200 nm to several hundred nanometers, and eventually formed highly ordered sheet-like nanostructures of micrometer size (Fig. 5a). This phenomenon gave further evidence of the feasibility of developing size-controllable high dimensional nanoarrays under photo-induced protein assembly (Fig. 5c). When we characterized the assemblies in TEM, detailed superstructures were presented clearly. Fig. 5b showed tightly packed, extended sheet-like architectures composed of regular repeating SP1S98Y units. If observed carefully, we can find that these SP1S98Y rings formed hexagonally packed nanosheets presenting a staggered arrangement, which could realize charge complementation in lateral directions to generate thermodynamically favorable products with minimum energy. All these data coincided with our prediction of manipulating covalent protein assembly based on the designed Tyr–Tyr coupling upon photocatalysis.
 | ||
| Fig. 5 (a) SEM images of the programmed assembly procedures with illumination for 15 min, 30 min, and 45 min, respectively; (b) TEM analysis of the assembled nanosheets. The inset is the detailed morphology and the corresponding scheme model; (c) schematic representation of the photocontrolled gradually increasing behavior. | ||
As we have demonstrated the effective cross-linking of the target Tyr residues in fabricating the desired nanostructures, a reasonable mechanism in this assembly system should also be explored. As shown in Scheme 2, the efficient visible light-harvesting catalyst Ru(bpy)32+ was known to produce an excited state under photolysis, which was able to donate an electron to persulfate for the cleavage of an O–O bond. Ru(III)-mediated formation of a tyrosyl radical was proposed as an initiating step. Coupling of this radical with the adjacent Tyr residues and subsequent removal of a hydrogen atom by the sulfate radical could accomplish the reaction, thus achieving protein assembly by Tyr–Tyr coupling. This oxidative coupling was an appealing strategy on which to base a photo-induced reaction because it not only resulted in the direct coupling of target residues without an intervening linker arm, but also can achieve precise control of the assembly morphologies.
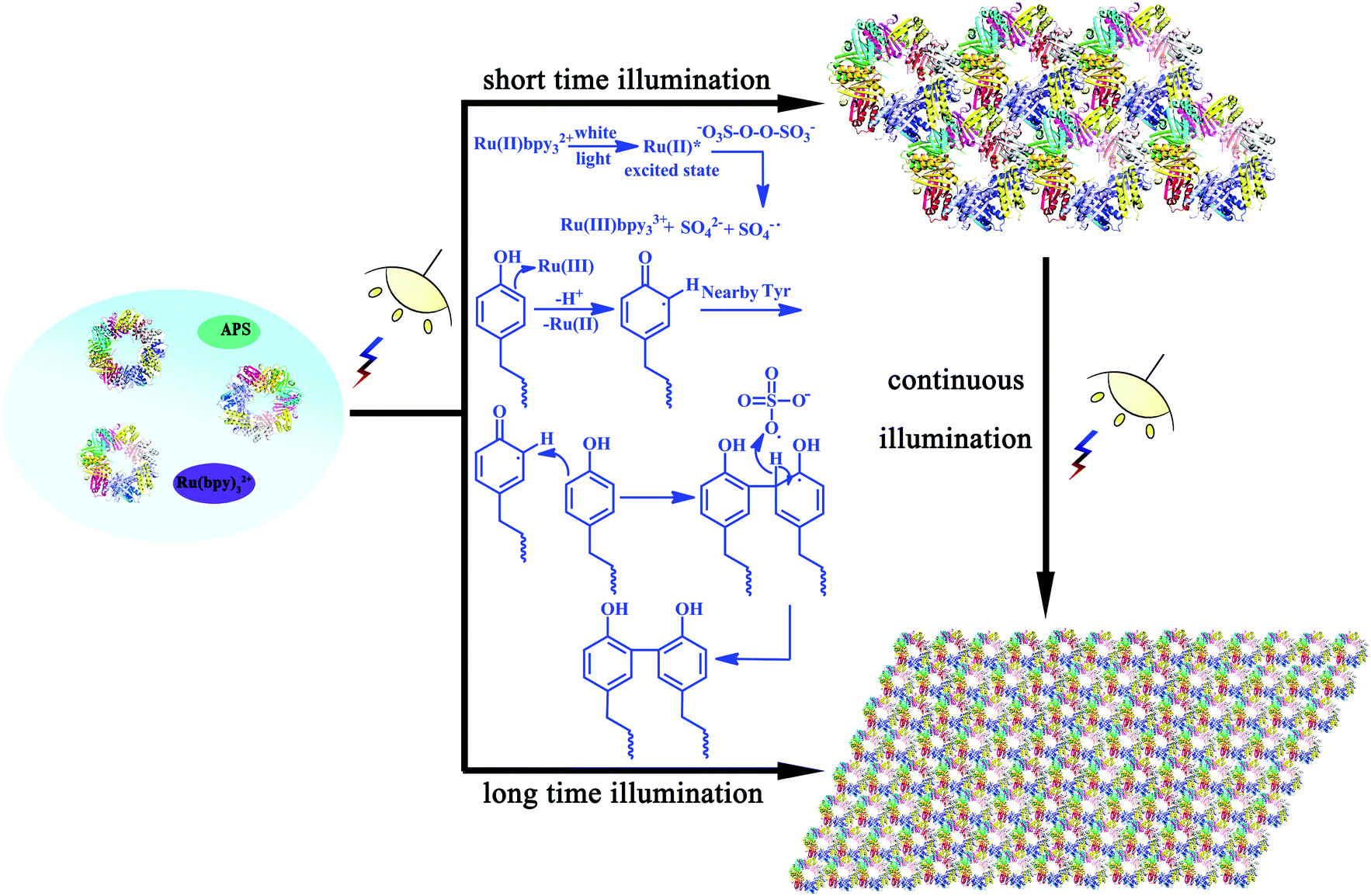 | ||
| Scheme 2 Plausible mechanism of the photo-induced protein assembly process and schematic representation of the photocontrolled assembly behavior. | ||
The outstanding predominance of this photo-induced strategy via covalent interactions was mainly reflected in the structural stability compared to those based on conventional weak interactions. In order to verify this phenomenon, heat treatment from 60 °C to 80 °C for 30 min was carried out for the assembly samples. AFM images gave strong evidence of the maintenance of the assembled sheet structures and the height of the architectures also agreed with the crystal structure of the model protein (Fig. S8a and b, ESI†). Surprisingly, we found that the nanosheets can keep the original assembled pattern even when the temperature was up to 80 °C, exhibiting excellent thermal stability (Fig. S8d, ESI†). This excellent feature endowed the self-assembled protein architectures with promising applications.
Conclusions
In summary, we successfully developed a facile photo-induced strategy for Ru(bpy)32+ driven covalent protein assembly. Combining computer simulation and specific coupling, by manipulating the stoichiometry of the photocatalysis and protein concentration, defined 2D protein architectures with a significantly ordered ring-shaped packing mode were constructed. The self-assembly process and detailed nanostructures have been studied by a series of characterizations including fluorescence spectroscopy, DLS, AFM, SEM, TEM, etc. By regulating the illumination time, the assembly system can achieve programmed growth procedures in fabricating photo-induced superstructures with precise size control. Notably, the constructed 2D nanosheets via covalent interactions also exhibited prominent structural stability, which facilitated their applications in practical fields. Following these dramatic features, we believe that such advanced architectures hold great promise in the development of protein-based nanobiomaterials.Experimental procedures
Construction, expression, and purification of SP1S98Y
Construction of pET22b plasmids containing SP1S98Y protein genes can be realized by site-directed mutagenesis. The primers were synthesized by Sangon Biotech and the plasmid pET22b-SP1 was used as the template for PCR cloning to obtain the mutant plasmids pET22b-SP1S98Y. The resulting plasmids were confirmed by DNA sequencing and then transformed into Escherichia coli BL21 (DE3) for protein expression. The E. coli BL21 (DE3) strains were cultured in Luria broth (LB) containing 100 μg mL−1 ampicillin with shaking at 37 °C. Then 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to the SP1S98Y LB when OD600 reached 0.8 to induce protein expression. The cells were incubated at 28 °C for 4 h and then harvested by centrifugation at 8000 rpm for 15 min. Resuspended in 20 mM Tris–HCl buffer (pH 8.0), the cells were sonicated to extract the protein. The insoluble pellets were separated by centrifugation at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min.
000 rpm for 30 min.
For purification, soluble mutant proteins SP1S98Y were first heated at 85 °C and centrifuged at 15000 rpm for 30 min. The supernatant was further purified by a DEAE ion-exchange column in 20 mM Tris–HCl buffer. The eluted solution was a gradient of NaCl from 100 to 500 mM, and the target protein was eluted in 500 mM NaCl. For further purification, the protein was loaded into a Sephadex G75 with 20 mM PBS buffer (pH 7.4). The obtained solution was dialyzed to remove the salts.
Photo-induced assembly of the 2D protein nanosheets
The assembly process was carried out in a total volume of 1 mL in a buffer consisting of 10 mM PBS (pH 7.0), 5 × 10−6 M Ru(bpy)32+ and freshly purified SP1S98Y (10−5 M). Ammonium persulfate (1.25 × 10−4 M) was added to the above solution just before irradiation. The solution was placed in a 1.5 mL Eppendorf tube positioned parallel to the beam of light at a distance of 20 cm from 60 W white-light sources. Exposure time was controlled manually. After photo-induced coupling, the assemblies were dialyzed in a dark place to remove the Ru(bpy)32+ and APS, thus to obtain the 2D nanosheets.DLS measurement
DLS measurement was performed on a Malvern Instrument Zetasizer Nano ZS instrument at 25 °C to study the hydrodynamic diameters of the SP1S98Y-based self-assemblies varying with time. SP1S98Y was dissolved in 10 mM pH = 7.0 PBS and the final concentration was 10 μM. Samples were made with appropriate concentrations of Ru(bpy)32+ and APS and were illuminated for different times prior to DLS measurement. The hydrodynamic diameters of the self-assemblies increase with time, which well confirmed the photocontrolled assembly mode.Fluorescence spectroscopy measurement
The fluorescent measurement was carried out on a Shimadzu RF-5301 PC spectrofluorimeter. Under the optimum experimental conditions, the excitation and emission wavelengths of the protein assemblies were 325 and 405 nm, respectively, and the fluorescence signal was recorded from 350–550 nm. Thus, the assembly process and photo-induced mechanism were identified and monitored using the fluorescence emission spectrum. As time went on, the ratio of protein assemblies became larger and ultimately approached a maximum, which can be reflected in the spectrum.AFM and SEM characterization
Samples for AFM and SEM characterization were prepared using the same method: dropping 10 μL of a 1 μM solution of protein assemblies on a freshly prepared hydroxylated silicon wafer for 10 min, washing with Milli-Q water, and drying under air before being employed in AFM/SEM. The AFM measurement was performed on a Nanoscope III controller (Veeco Metrology, Santa Barbara, CA, USA) and the SEM observations were carried out on a JEOL FESEM 6700F scanning electron microscope where the primary electron energy was 3.00 kV.TEM characterization
The TEM images were recorded by a JEM-2100F instrument with an accelerating voltage of 200 kV. Samples were adsorbed on Formvar carbon-coated copper grids for 10 min and then drawn away by pipet. After being negatively stained by adding 4 μL of stain (2% sodium phosphotungstate in Milli-Q water) onto the grid for 40 s, the excess strain was removed. The samples were dried under air flow overnight before imaging.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Prof. Oded Shoseyov from The Hebrew University of Jerusalem for his kind supply of the SP1 gene. This work was also supported by the National Natural Science Foundation of China (No. 21234004, 21420102007, 21574056, and 91527302), the Chang Jiang Scholars Program of China and the Graduate Innovation Fund of Jilin University (2017078).Notes and references
- J. A. Marsh, H. Hernández, Z. Hall, S. E. Ahnert, T. Perica, C. V. Robinson and S. A. Teichmann, Cell, 2013, 153, 461–470 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- S. E. Ahnert, J. A. Marsh, H. Hernández, C. V. Robinson and S. A. Teichmann, Science, 2015, 350, aaa2245 CrossRef PubMed.
- Y. S. Bai, Q. Luo and J. Q. Liu, Chem. Soc. Rev., 2016, 45, 2756–2767 RSC.
- H. C. Sun, Q. Luo, C. X. Hou and J. Q. Liu, Nano Today, 2017, 14, 16–41 CrossRef CAS.
- G. Yang, L. B. Wu, G. S. Chen and M. Jiang, Chem. Commun., 2016, 52, 10595–10605 RSC.
- N. P. King, J. B. Bale, W. Sheffler, D. E. McNamara, S. Gonen, T. Gonen, T. O. Yeates and D. Baker, Nature, 2014, 510, 103–108 CrossRef CAS PubMed.
- Q. Luo, C. X. Hou, Y. S. Bai, R. B. Wang and J. Q. Liu, Chem. Rev., 2016, 116, 13571–13632 CrossRef CAS PubMed.
- S. Jin, D. D. Zheng, B. Sun, X. H. Yu, X. Zha, Y. J. Liu, S. M. Wu and Y. Q. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 34244–34251 CAS.
- S. Y. Qin, Y. Pei, X. J. Liu, R. X. Zhuo and X. Z. Zhang, J. Mater. Chem. B, 2013, 1, 668–675 RSC.
- T. P. Fu, P. A. Guerette, R. Y. T. Tan, H. Zhao, L. Schefer, R. Mezzenga and A. Miserez, J. Mater. Chem. B, 2015, 3, 2671–2684 RSC.
- M. Künzle, T. Eckert and T. Beck, J. Am. Chem. Soc., 2016, 138, 12731–12734 CrossRef PubMed.
- L. L. Zhao, S. P. Qiao and J. Q. Liu, Sci. China: Chem., 2016, 59, 1531–1540 CrossRef CAS.
- G. Yang, H. M. Ding, Z. Kochovski, R. T. Hu, Y. Lu, Y. Q. Ma, G. S. Chen and M. Jiang, Angew. Chem., Int. Ed., 2017, 56, 10691–10695 CrossRef CAS PubMed.
- L. Z. Ma, F. Li, T. Fang, J. Zhang and Q. B. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 11024–11031 CAS.
- C. Ma, X. M. Liu, G. Y. Wu, P. Zhou, Y. T. Zhou, L. Wang and X. Huang, ACS Macro Lett., 2017, 6, 689–694 CrossRef CAS.
- G. D. Han, J. T. Wang, X. T. Ji, L. Liu and H. Y. Zhao, Bioconjugate Chem., 2017, 28, 636–641 CrossRef CAS PubMed.
- L. Miao, J. S. Han, H. Zhang, L. L. Zhao, C. Y. Si, X. Y. Zhang, C. X. Hou, Q. Luo, J. Y. Xu and J. Q. Liu, ACS Nano, 2014, 8, 3743–3751 CrossRef CAS PubMed.
- H. C. Sun, L. Miao, J. X. Li, S. Fu, G. An, C. Y. Si, Z. Y. Dong, Q. Luo, S. J. Yu, J. Y. Xu and J. Q. Liu, ACS Nano, 2015, 9, 5461–5469 CrossRef CAS PubMed.
- V. Liljeström, J. Seitsonen and M. A. Kostiainen, ACS Nano, 2015, 9, 11278–11285 CrossRef PubMed.
- J. Mikkilä, E. Anaya-Plaza, V. Liljeström, J. R. Caston, T. Torres, A. de la Escosura and M. A. Kostiainen, ACS Nano, 2016, 10, 1565–1571 CrossRef PubMed.
- J. D. Brodin, X. I. Ambroggio, C. Y. Tang, K. N. Parent, T. S. Baker and F. A. Tezcan, Nat. Chem., 2012, 4, 375–382 CrossRef CAS PubMed.
- Y. Suzuki, G. Cardone1, D. Restrepo, P. D. Zavattieri, T. S. Baker and F. A. Tezcan, Nature, 2016, 533, 369–373 CrossRef CAS PubMed.
- S. P. Qiao, C. Lang, R. D. Wang, X. M. Li, T. F. Yan, T. Z. Pan, L. L. Zhao, X. T. Fan, X. Zhang, C. X. Hou, Q. Luo, J. Y. Xu and J. Q. Liu, Nanoscale, 2016, 8, 333–341 RSC.
- R. P. G. Bosmans, J. M. Briels, L. Milroy, T. F. A. de Greef, M. Merkx and L. Brunsveld, Angew. Chem., Int. Ed., 2016, 128, 9045–9049 CrossRef.
- P. J. de Vink, J. M. Briels, T. Schrader, L. Milroy, L. Brunsveld and C. Ottmann, Angew. Chem., Int. Ed., 2017, 56, 1–6 CrossRef PubMed.
- C. X. Hou, J. X. Li, L. L. Zhao, W. Zhang, Q. Luo, Z. Y. Dong, J. Y. Xu and J. Q. Liu, Angew. Chem., Int. Ed., 2013, 125, 5700–5703 CrossRef.
- E. R. Ballister, A. H. Lai, R. N. Zuckermann, Y. F. Cheng and J. D. Mougous, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3733–3738 CrossRef CAS PubMed.
- H. C. Sun, X. Y. Zhang, L. Miao, L. L. Zhao, Q. Luo, J. Y. Xu and J. Q. Liu, ACS Nano, 2016, 10, 421–428 CrossRef CAS PubMed.
- L. Miao, Q. S. Fan, L. L. Zhao, Q. L. Qiao, X. Y. Zhang, C. X. Hou, J. Y. Xu, Q. Luo and J. Q. Liu, Chem. Commun., 2016, 52, 4092–4095 RSC.
- J. F. Matthaei, F. DiMaio, J. J. Richards, L. D. Pozzo, D. Baker and F. Baneyx, Nano Lett., 2015, 15, 5235–5239 CrossRef CAS PubMed.
- X. Gao, S. Yang, C. C. Zhao, Y. H. Ren and D. Z. Wei, Angew. Chem., Int. Ed., 2014, 126, 1–5 CrossRef.
- Y. S. Bai, Q. Luo, W. Zhang, L. Miao, J. Y. Xu, H. B. Li and J. Q. Liu, J. Am. Chem. Soc., 2013, 135, 10966–10969 CrossRef CAS PubMed.
- H. Kitagishi, Y. Kakikura, H. Yamaguchi, K. Oohora, A. Harada and T. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 1271–1274 CrossRef CAS PubMed.
- P. Ringler and G. E. Schulz, Science, 2003, 302, 106–109 CrossRef CAS PubMed.
- M. A. Kostiainen, P. Ceci, M. Fornara, P. Hiekkataipale, O. Kasyutich, R. J. M. Nolte, J. J. L. M. Cornelissen, R. D. Desautels and J. van Lierop, ACS Nano, 2011, 5, 6394–6402 CrossRef CAS PubMed.
- N. Kong, Q. Peng and H. B. Li, Adv. Funct. Mater., 2014, 24, 7310–7317 CrossRef CAS.
- H. C. Sun, L. L. Zhao, T. T. Wang, G. An, S. Fu, X. M. Li, X. L. Deng and J. Q. Liu, Chem. Commun., 2016, 52, 6001–6004 RSC.
- A. Heyman, I. Medalsy, O. B. Or, O. Dgany, M. Gottlieb, D. Porath and O. Shoseyov, Angew. Chem., Int. Ed., 2009, 48, 1–6 CrossRef.
- I. Medalsy, O. Dgany, M. Sowwan, H. Cohen, A. Yukashevska, S. G. Wolf, A. Wolf, A. Koster, O. Almog, I. Marton, Y. Pouny, A. Altman, O. Shoseyov and D. Porath, Nano Lett., 2008, 8, 473–477 CrossRef CAS PubMed.
- L. L. Zhao, H. Y. Zou, H. Zhang, H. C. Sun, T. T. Wang, T. Z. Pan, X. M. Li, Y. S. Bai, S. P. Qiao, Q. Luo, J. Y. Xu, C. X. Hou and J. Q. Liu, ACS Nano, 2017, 11, 938–945 CrossRef CAS PubMed.
- D. A. Fancy and T. Kodadek, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6020–6024 CrossRef CAS.
- J. Fang, A. Mehlich, N. Koga, J. Q. Huang, R. Koga, X. Y. Gao, C. G. Hu, C. Jin, M. Rief, J. Kast, D. Baker and H. B. Li, Nat. Commun., 2013, 4, 2974–2984 Search PubMed.
- B. Tang and Y. Wang, Spectrochim. Acta, Part A, 2003, 59, 2867–2874 CrossRef.
- B. Tang, Y. Wang, H. L. Liang, Z. Z. Chen, X. W. He and H. X. Shen, Spectrochim. Acta, Part A, 2006, 63, 609–613 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, circular dichroism spectra, dynamic light scattering, and additional AFM and TEM images. See DOI: 10.1039/c7tb02826a |
| This journal is © The Royal Society of Chemistry 2018 |