
The influence of bubble size on chondrogenic differentiation of adipose-derived stem cells in gelatin microbubble scaffolds†
Kuan-Han
Wu‡
a,
Chieh
Mei‡
a,
Che-Wei
Lin
a,
Kai-Chiang
Yang
 b and
Jiashing
Yu
*a
b and
Jiashing
Yu
*a
aDepartment of Chemical Engineering, College of Engineering, National Taiwan University, Taipei 106, Taiwan. E-mail: jiayu@ntu.edu.tw
bCollege of Medicine, Taipei Medical University, Taipei 110, Taiwan
First published on 22nd November 2017
Abstract
In human bodies, cartilage tissue lacks the ability to heal when it encounters trauma or lesions. This inability of cartilage tissue to self-repair motivates all sorts of studies on autologous chondrocyte transplantation; however, the drawback of high chondrocyte concentration is hard to overcome due to the loss of differentiated chondrocyte phenotype during cell culture. The differentiation of stem cells into chondrocytes is a possible solution to provide a large number of differentiated chondrocytes. In this study, human adipose-derived stem cells (hASCs) have been chosen as a model for further differentiation into chondrocytes. Studies on the influence of porous biomaterials on cell behavior have been performed to determine the best conditions for stem cell differentiation. Among these conditions, bubble or pore size is a factor that is commonly discussed. In our study, we fabricated four gelatin microbubble scaffolds with different pore sizes, but uniform spherical shapes by microfluidic techniques. Then, we compared the influence of pore size on cell growth and differentiation. Previously, we have examined adipogenesis and osteogenesis of hASCs in this scaffold. In this study, we focused on the influence of pore size on chondrogenesis. According to the experimental results of immunofluorescence staining, GAG content, and qPCR, the largest pore size, which is 200 μm in diameter, shows the best chondrogenesis result.
1. Introduction
Articular cartilage lacks the ability of spontaneous healing when damaged.1 Therefore, when cartilage defects occur, they may further lead to osteoarthritis (OA). OA is a degenerative joint disease, and 80% of patients with OA will have limitations in their movements. Moreover, according to the World Health Organization, 9.6% of men and 18% of women over the age of 60 have symptomatic OA. Many researchers have studied treatments for cartilage defects.For the treatment of cartilage defects, autologous chondrocyte implantation was first used in clinical practice.2Via the biopsy of healthy cartilage, researchers have cultured isolated chondrocytes and injected them into the damaged cartilage. Large amounts of chondrocytes are required during implantation. However, these chondrocytes have limited proliferation ability and lose their functional phenotype during culture. Therefore, treatments substituting chondrocytes with mesenchymal stem cells have been an alternative method studied recently.3,4
To induce chondrogenic differentiation into stem cells, many physical factors, such as stiffness, topography, and pore size, have been taken into consideration during culture;5 considering stiffness, different tissues in the human body have a variety of stiffness levels. Fibrous cartilage tissue possesses an elasticity of around 20 kPa.6 An ideal culturing environment should have similar elasticity as the targeted tissue in the human body.7–9 With regard to topography, studies have shown that the differentiation of stem cells can be manipulated with different micropatterned surfaces including grooves,10,11 nanodot arrays,12 pillars,13 and all sorts of different shapes at the interface.14 With regard to pore size, studies on polycaprolactone (PCL) scaffolds showed that stem cells displayed different morphologies after differentiation when cultured on scaffolds with different pore sizes. Between the pore sizes of 100 and 200 μm, adipose-derived stem cells (ASCs) proliferated and spread on the surface, whereas at 400 μm, there were lumps of aggregated ASCs. This showed the influence of pore size on ASC differentiation.15 Among these factors, pore size has been studied the least due to the presence of heterogeneous pore sizes in most substrates.16–18 In previous studies, scaffolds have been made through freeze-drying or gas foaming methods to obtain substrates with various pore sizes.19 The investigation of pore size was not accurate enough. In this study, a microfluidic device was applied to fabricate scaffolds with homogeneous pore sizes and further study the influence of pore size on chondrogenic differentiation of hASCs (Fig. 1).
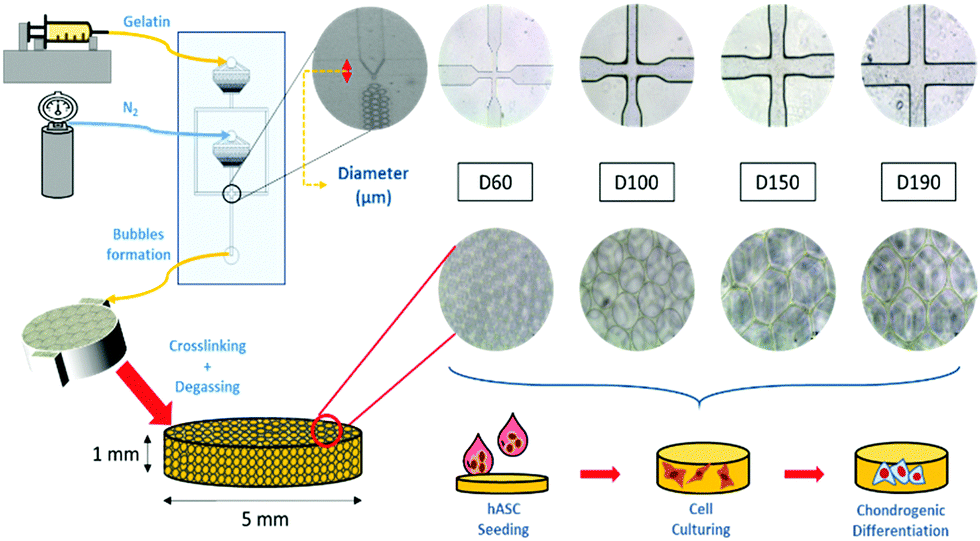 | ||
| Fig. 1 Schematic of gelatin microbubble scaffold fabrication. | ||
2. Materials and methods
2.1 Scaffold fabrication
A more detailed description of the process has been provided in previous studies.20,21 In brief, our microfluidic device was made from a customized silicon wafer with a polydimethylsiloxane (Silmore) mold through the soft lithography method. Each microfluidic channel has two inputs, for liquid and gas, and one output for bubbles. The liquid input is composed of 7% gelatin (SIGMA, Lot# 041M0052V) and 1% Pluronic® F127 (SIGMA, CAS Number 9003-11-6) surfactant, and the gas input is mainly composed of N2 (FMI) with some amount of perfluorohexane (Synquest Laboratories, CAS# 355-42-0). After the liquid enters the channel, it branches into two and intersects with the gas flow at a cross-section (width = 60, 100, 150, and 190 μm). The width of the cross-section affects the size of the bubbles formed.22 With a larger cross-section width, larger bubbles are formed. With an adequate liquid flow rate and gas pressure, bubbles form and flow out. In this study, the liquid flow rate was controlled in the range from 40 to 60 μl min−1, and the air pressure was at 15 to 25 psi. Once stable bubbles were formed, they were then collected into pre-cooled cylindrical reservoirs with a diameter of 5 mm and height of 1 mm, which was the size of the scaffolds. After collection, the reservoirs were placed in a refrigerator at 4 °C for solidification of the gelatin microbubbles for 30 min. Then, the solidified gelatin microbubbles were crosslinked with 2% glutaraldehyde (SIGMA-ALDRICH, Lot# MKBD9857) in a refrigerator at 4 °C overnight. To generate interconnected pores among bubbles, a degas treatment was conducted by placing reservoirs in vacuum for 2 hours. Furthermore, we eliminated the autofluorescence of gelatin by soaking these scaffolds in a 0.5% sodium borohydride (ALDRICH) solution for 1 hour. Finally, they were stored in a 1% penicillin/streptomycin/amphotericin solution at 4 °C before use. Between each step, the scaffolds were rinsed thoroughly with PBS buffer.2.2 Characterization of the gelatin microbubble scaffold
Uniformity of the scaffolds was observed using an inverted microscope and the pore size of the scaffolds was measured using a scanning electron microscope (SEM; JSM-5310, JEOL). For SEM pre-treatments, scaffolds were frozen at −20 °C overnight and then freeze-dried. The freeze-dried samples were attached to the carrier and then electroplated with gold. To investigate the mechanical properties of the scaffold, a compression test was performed to calculate the Young's modulus of the scaffold. The measurement was conducted using the elastic modulus load cells LTS-200GA (KYOWA, Japan) and a Stepping Motor Driven Stages SGSP (MS) 20-85 (SIGMA KOKI, Japan). Normal stress on the axial direction was measured, and strain was calculated to obtain the Young's modulus.23 The mechanical properties, including elasticity and stiffness, of the scaffolds were investigated and measured by producing a stress-to-strain diagram. The diagram could be divided into four zones, which represented different states during the compression process, and the Young's moduli were calculated by the slopes in zone I and zone III. Zone I indicated the elasticity of the scaffold structure, whereas zone III indicated the stiffness of the material itself. These two Young's moduli were calculated for each group of scaffolds.2.3 hASC culture and seeding
Herein, hASC (National Taiwan University Hospital) was cultured in a DMEM-HG/F12 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) medium with 1 ng L−1 Human FGF, 10% (v/v) FBS, and penicillin (100 IU per ml). Cells were cultured at 37 °C under 5% CO2, and the medium was renewed every two days until confluence was reached. To remove cells from the tissue culture polystyrene (TCPS), after PBS washing, trypsin–EDTA (Biological Industries) was added to TCPS and reacted at 37 °C for 5 min. To stop the enzyme reaction, the culture medium was added to the dish, and this was followed by centrifugation. After removing the supernatant, an adequate amount of the culture medium was added to the centrifuged cells for re-suspension. The number of suspended cells was counted using a hemocytometer; cells were stained with trypan blue to test the cell viability. Before seeding, all the samples were immersed in 70% ethanol for 30 min and then in PBS before use. For scaffold seeding, the liquid in scaffolds was removed by sterilized filter paper. After this, around 15 μl of condensed cell suspension that contained around 38000 cells was dropped on the scaffolds. To prevent cells from detaching, the well plates were incubated for more than 2 hours before adding the fresh medium. For 2D control, the fresh medium was added before the addition of the cell suspension to provide a homogeneous cell distribution condition. For chondrogenic differentiation, the growth medium was substituted with the induction medium after 1 day of incubation. The chondrogenic induction medium was composed of DMEM/HG with 1% (v/v) FBS, 10 ng ml−1 TGF-β1, 6.25 μg ml−1 insulin, 50 μM ascorbate-2-phosphate, and penicillin (100 IU per ml). The induction medium was also refreshed every two to three days.
:1) medium with 1 ng L−1 Human FGF, 10% (v/v) FBS, and penicillin (100 IU per ml). Cells were cultured at 37 °C under 5% CO2, and the medium was renewed every two days until confluence was reached. To remove cells from the tissue culture polystyrene (TCPS), after PBS washing, trypsin–EDTA (Biological Industries) was added to TCPS and reacted at 37 °C for 5 min. To stop the enzyme reaction, the culture medium was added to the dish, and this was followed by centrifugation. After removing the supernatant, an adequate amount of the culture medium was added to the centrifuged cells for re-suspension. The number of suspended cells was counted using a hemocytometer; cells were stained with trypan blue to test the cell viability. Before seeding, all the samples were immersed in 70% ethanol for 30 min and then in PBS before use. For scaffold seeding, the liquid in scaffolds was removed by sterilized filter paper. After this, around 15 μl of condensed cell suspension that contained around 38000 cells was dropped on the scaffolds. To prevent cells from detaching, the well plates were incubated for more than 2 hours before adding the fresh medium. For 2D control, the fresh medium was added before the addition of the cell suspension to provide a homogeneous cell distribution condition. For chondrogenic differentiation, the growth medium was substituted with the induction medium after 1 day of incubation. The chondrogenic induction medium was composed of DMEM/HG with 1% (v/v) FBS, 10 ng ml−1 TGF-β1, 6.25 μg ml−1 insulin, 50 μM ascorbate-2-phosphate, and penicillin (100 IU per ml). The induction medium was also refreshed every two to three days.
2.4 LIVE/DEAD assay
To evaluate cell survival within the 3D scaffolds, the seeded hASC cells were stained using the LIVE/DEAD® Cell Imaging Kit (Invitrogen) (N = 3) after 1 day of culture. At first, the medium in each sample was removed. Then, after rinsing in PBS, the samples were immersed in a solution with 2 μM calcein AM and 4 μM ethidium homodimer-1 (EthD-1) in PBS for 45 minutes. Calcein AM was enzymatically converted into calcein with green fluorescence, and the EthD-1 could only pass through the incomplete cell membrane of dead cells and bind with nucleic acid; this caused red fluorescence. After staining, the results were observed using a fluorescence microscope.2.5 Cell viability assay
To calculate cell proliferation and cell viability in the scaffolds, 3 × 104 cells per sample were seeded to both 2D control and 3D scaffolds. After 1, 4, and 7 days of culture, the samples were washed with PBS and further cultured in a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) working solution for 3.5 hours. After this, the MTT working solution was replaced with DMSO to dissolve the purple crystals formed during culture with the MTT working solution and shaken for 30 min. After shaking, 100 μl of the DMSO solution of samples was moved to a 96-well plate. The optical density value (OD) at 492 nm was measured by an ELISA reader. The pure DMSO solution was used as the blank solution. The OD value of each group was compared with that of every other group to analyze the relative cell activity (N = 3).2.6 Nucleus and F-actin labelling
Due to the multiple-layer structure of the 3D porous scaffolds, only the cells on the top layer can be observed without fluorescent staining. Therefore, aside from optical microscope observation, cell morphology was further observed using fluorescent staining. After 1, 4, and 7 days of culture, the samples were washed with PBS. Next, the cells were fixed with 3.7% formaldehyde for 10 minutes at room temperature and then soaked in 0.2% Triton-X 100 for 10 minutes to permeabilize them. After this, the samples were stained with phalloidin (SIGMA), which was diluted 1:100 with PBS. Then, the phalloidin dye was replaced with DAPI (SIGMA), which was diluted 1:500 with DI water. Between every step, PBS washing was required. Before observation, each of the samples was washed with PBS. Images of the cells in the scaffolds or on the 2D controls were obtained by a confocal microscope and inverted fluorescence microscope (N = 3).
2.7 Immunofluorescence staining
The chondrogenic differentiation results of hASCs were tested by immunofluorescence staining. After 14 and 21 days of culture in the induction medium, cells were first fixed and permeabilized. To prevent non-specific binding, a blocking procedure was required. Cells were immersed in a 10% fetal bovine serum solution in PBST. After being blocked in the PBST buffer, the samples were incubated with two primary antibodies, anti-COLII and anti-SOX9, overnight at 4 °C. The primary antibodies were diluted with the blocking buffer at 1:200. Following primary antibody incubation, the samples were immersed in a Alexa Fluor® 488-conjugated AffiniPure Goat Anti-Rabbit IgG secondary antibody solution for two hours followed by rinsing for three times. Finally, the images of each sample were obtained using a confocal microscope after nucleus labeling (N = 3).
2.8 Dimethylmethylene blue (DMMB) assay
To quantify the chondrogenic ability of the hASCs with different pore sizes, a more specific test was required. Therefore, we chose the DMMB assay to quantify the amount of glycosaminoglycans in each group after differentiation as a marker of chondrogenic ability. After 14 and 21 days of differentiation, the samples were washed with PBS and further digested with a 125 μg ml−1 papain solution at 60 °C overnight. The papain powder was dissolved in the PBE buffer. The PBE buffer was consisted of 100 mM disodium phosphate and 5 mM EDTA. The next day, the digested solution was obtained in an Eppendorf and preserved at −20 °C until use. Before performing the experiment, the DMMB solution was prepared with 40 mM sodium chloride, 40 mM glycine, and 46 μM DMMB. The pH of the DMMB solution was adjusted to 3.0 using a hydrochloride solution. In a 96-well plate, 50 μl of the digested sample and 200 μl the DMMB solution were mixed. Chondroitin sulfate was used as the control standard. After mixing, absorbance at 525 nm was measured using an ELISA plate reader24,25 (N = 3).2.9 RNA extraction and qPCR
Samples were washed with PBS and immersed in REzolTM C&T (rezol); the samples were shaken for 5 min to achieve full extraction. The solution was moved to a micro-centrifuge tube, vortexed, and kept in 4 °C for 5 min. Then, chloroform with 1/5 volume of the solution was added, and the solution was vortexed and kept at 4 °C for 5 min again. The tubes were centrifuged at 14000 rpm for 5 min at 4 °C. The supernatant layer was placed in a new micro-centrifuge tube. Next, the same volume of isopropanol was added to the tube and kept at −20 °C overnight for precipitation. The next day, the samples were centrifuged at 14000 rpm at 4 °C for 45 min. Isopropanol was then removed, and the RNA pellet was left at the bottom. The pellet was washed with 75% ethanol. Ethanol was removed after centrifugation. Finally, 20 μl of DI water was added to re-dissolve the RNA pellet. The concentration of RNA was measured by the Nanodrop machine. The solution was stored at −20 °C.
Extracted RNA is the template of complementary DNA (cDNA) in reverse transcription (RT). RNA with a known concentration was mixed with RNase-free water to reach the same concentration for each group. The concentration was around 5 μg in 10 μl, and the solution was heated to 65 °C before reaction. Then, 10 μl of RT master mix and 1 μl of RNase inhibitor were added to the solution for further reaction. The reaction conditions were adjusted by the PCR machine: 50 °C for 60 min, 70 °C for 5 min, and then cooling to 4 °C for 5 min. The final DNA sample was stored at 4 °C.
The gene expressions of different groups were compared using real time PCR, also known as qPCR, with amplification of target gene by fluorescence signal during the process. The fluorescent-labelled dye SYBR Green was used to quantitate the PCR products. DNA primers in the experiments were diluted with DNase/RNase-free water to three times the volume.
During the cycle of temperature changes, the targeted DNA sequence was copied and multiplied, and the fluorescence signal was also amplified. When a threshold of the fluorescence signal was reached, the number of cycles completed was determined. With these values, the original amount of the targeted DNA sequence was measured and compared with that for the different groups (N = 3).
2.10 Statistical analysis
All data were expressed as means standard deviation. Comparison of different groups was performed using the Student's t-test, and significant difference was considered when the P value ≤0.05. The statistical data was analyzed using the Microsoft Excel Software (version 2010).3. Results and discussions
3.1 Scaffold characterization
After fabrication of the scaffolds, the structure and pore size of the four scaffolds were observed via SEM (Fig. 2(a)–(d)) and optical reversal microscopy images (Fig. 2(e)–(h)). The optical microscopy images show that each scaffold was consisted of microbubbles of similar shape and size. This allowed us to control the variable among each group to pore size only. As seen in the SEM image, the microbubbles were interconnected after degassing. Therefore, the degassing process generated pores for cells to penetrate the surface of the scaffolds instead of growing only on the surface. As abovementioned in the previous section, there were four different diameters of cross-section in the microfluidic device used to generate microbubbles: D60, D100, D150, and D190. In general, as the diameter of the cross-section increased, the resulting bubble size increased. However, an upper limit of the bubble size was observed as the diameter of the cross-section reached 150 μm. This result is induced by the fact that the diameter of output channel is smaller than the diameter of the cross section. The bubble sizes were measured by the built-in calculating system of the SEM. Herein, we defined bubble size as the outer diameter of the chamber. According to the measurement in the SEM image, the bubble sizes of the four scaffolds were around 70, 110, 210, and 160 μm. Further research was performed based on these four groups of scaffolds.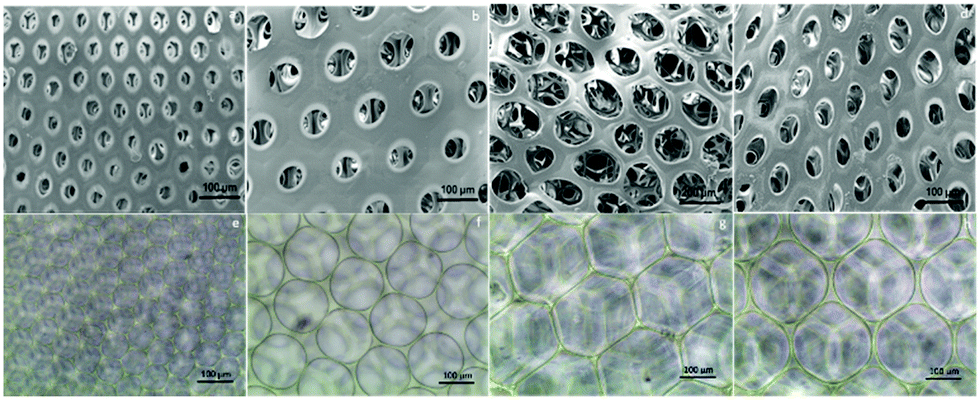 | ||
| Fig. 2 (a)–(d) The SEM images of the scaffolds with different pore sizes show that the 3D gelatin microbubble scaffolds are composed of numerous homogeneous microbubbles. (e)–(h) The corresponding optical microscopy image of the microbubble scaffolds in the SEM image. | ||
After the compression test of the scaffold, a stress–strain curve of each scaffold was obtained. According to the non-linear stress–strain curve abovementioned in the Materials and methods section.
The Young's modulus of the scaffold was calculated for zone I and zone III. In zone I, the Young's modulus indicated the elasticity of the scaffold structure. As shown in Fig. 3, the elasticity of all four groups lay in the range from 10000 Pa to 30000 Pa, which is similar to the elasticity of the fibrous cartilage tissue. Moreover, the D60, D100, and D150 groups had no significant difference in elasticity. Although D190 group had a smaller elasticity, its elasticity was still close to the elasticity of the cartilage tissue. Therefore, the scaffolds that we fabricated herein should be suitable for chondrogenic differentiation. For zone III, the Young's modulus indicated the stiffness of the material itself. Fig. 3b shows that the stiffness of the smaller pore-size groups D60 and D100 is lower as compared to that of the larger pore-size groups. This is possibly because a larger pore size leads to a more complete crosslinking with glutaraldehyde during the fabrication process. However, all 4 groups exhibited the same degree of stiffness.
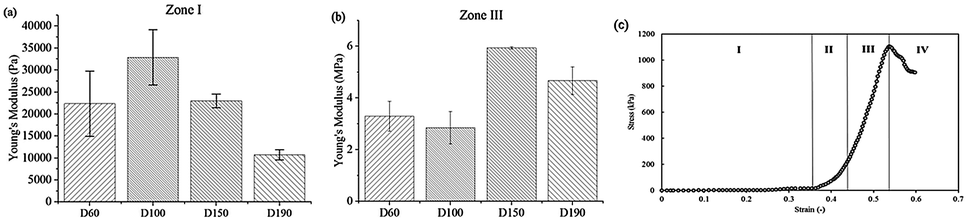 | ||
| Fig. 3 Young's modulus of the scaffold determined from the stress–strain curve obtained during the compression test in (a) zone I (b) zone III. (c) The stress–strain curve for porous scaffolds during the compression test. The curve can be divided into four parts. | ||
3.2 Cell survival and viability
To evaluate the cytotoxicity of the scaffolds, LIVE/DEAD staining was carried out to assess cell viability after culturing the cells in scaffolds for 1 day. In Fig. 4a–e, green fluorescence indicates live cells and red fluorescence indicates dead cells. In 2D groups, hASCs were uniformly spread on the surface and had only a few dead cells in comparison with live cells. Among the scaffold groups, D60 and D100 also had a few dead cells, whereas the D150 and D190 groups had even fewer dead cells; this result was likely because of the less efficient diffusion of oxygen and nutrients via pores with smaller sizes. However, cell viability was over 90% in each group. This indicates a high cell viability and low cytotoxicity for our scaffolds. The crosslinking agent glutaraldehyde used in scaffold fabrication is cytotoxic to cells. With these results, we could confirm that the unreacted crosslinking agent successfully reacted and was removed through washing and sodium borohydride reacting procedures. Moreover, we observed that in the scaffolds, there were signals from different layers of the scaffolds, which indicated that cells were alive inside the microbubbles. This corresponded to the fact that our microbubble scaffolds were multi-layered and interconnected, as observed from the SEM images.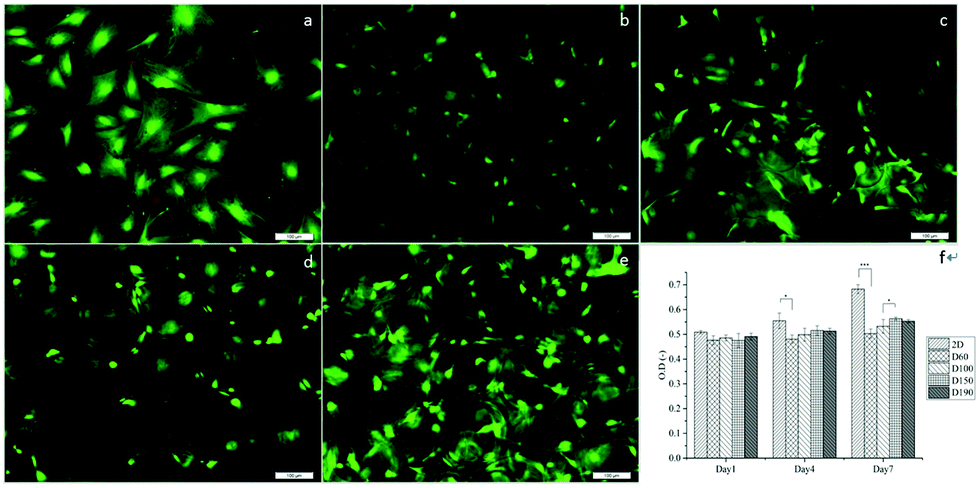 | ||
| Fig. 4 (a) Live/dead staining to 2D monolayers and (b) D60, (c) D100, (d) D150, and (e) D190 for cells cultured in 3D scaffolds with different pore sizes. Green fluorescence represents live cells, whereas red fluorescence indicates dead cells. Scale bar = 100 μm. (f) MTT assay revealed that cells had good viability within the microbubble scaffolds. *p < 0.05. ***p < 0.001. | ||
MTT assay was used to examine the cell activity and proliferation in the scaffolds. Fig. 4f shows that at day 1, the cell activity of every group was similar mostly due to the same amount of hASCs seeded and high cell viability. However, after day 4, the OD value of the 2D groups was significantly higher as compared to that of the 3D groups; this suggested a higher proliferation rate in the 2D groups; among the 3D groups, the OD values of the D150 and D190 groups were higher as compared to those of the D60 and D100 groups. In other words, scaffolds with larger pore sizes presented a higher proliferation rate. The possible reason for this phenomenon could be attributed to the seeding number of the cells. The seeding density was calculated in a previous study to reach confluence in the D60 groups. This suggested that in the D60 and D100 groups, there might not be extra space for cells to proliferate. In contrast, the D150 and D190 groups with larger pore sizes provide extra space for cells to proliferate.
3.3 hASC morphology
To start the investigation of the hASC behavior in scaffolds with various pore sizes, an inverted microscope was used to observe the hASC morphology directly. Fig. 5a, e and i show that in the D60 groups, hASCs present a more round shape. Aside from the round-shaped hASCs, there were some cells that elongated through adjacent microbubbles. This indicated that the microbubbles were interconnected, as observed in the SEM images.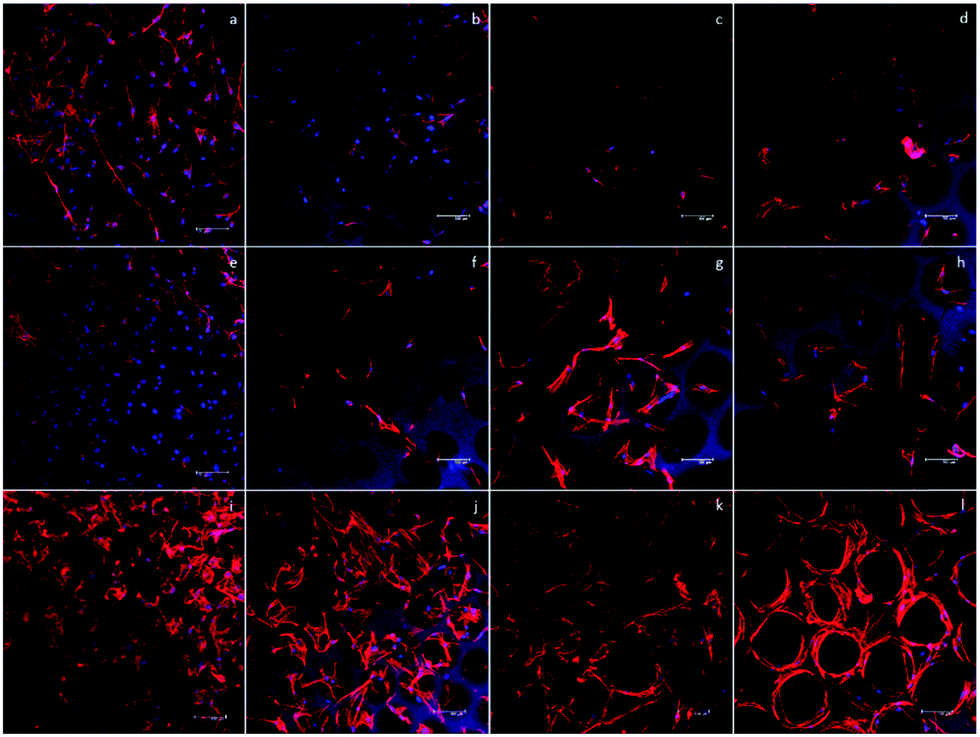 | ||
| Fig. 5 Nucleus and F-actin staining of the hASC cells in all 4 scaffolds for (a)–(d) day 1, (e)–(g) day 4, and (i)–(l) day 7. F-actin distribution was round in smaller pore size, whereas it was more elongated in larger pore sizes. Scale bar = 100 μm. | ||
In each D60 microbubble, the small bubble size allowed only one or two cells to grow. In the D100 group, a much lower ratio of the round cells was observed. Instead, most cells presented an elongated morphology, either crossing through adjacent bubbles or growing in a single bubble. In addition, there was a slightly higher number of cells growing in each microbubble. As the bubble size increased, more cells were observed in a single bubble. In the D150 and D190 groups, this trend could be observed clearly. In larger bubble groups, some hASCs presented an elongated morphology in a single bubble, whereas some hASCs grew around the stent. This was possibly due to the low curvature that made the microenvironment more similar to a flat plane and the extremely large radius that led to a long distance for cells to elongate across a single bubble.
However, due to the multi-layer structure of the microbubble scaffolds, only the first layer of the scaffolds could be observed without severe interference. In addition, the transparency of the scaffolds decreased as the bubble size decreased; this made the observation of the D60 groups more difficult than that of the others. Therefore, further fluorescence staining using phalloidin and DAPI to label F-actin and nuclei was carried out to have a closer observation of the morphology of the hASCs in the scaffolds.
Fig. 5 shows the fluorescence image of F-actin/DAPI staining after 1, 4, and 7 days of incubation. In addition, at day 7, we could observe that in D100, D150, and D190 groups, spatial distribution of hASCs was restricted by the bubble sphere. In the D190 group, hASCs even presented a hollow sphere-like shape. This sphere-like morphology was suggested to enhance the chondrogenic ability of the hASCs; this led to our following chondrogenic differentiation study.
3.4 Immunofluorescence staining
To qualitatively confirm the results of chondrogenic differentiation, immunofluorescence staining was conducted. There were two antibodies used: transcription factor SOX9 (SOX9) and collagen II (COLII). SOX9 has a dominant role in promoting chondrogenic differentiation by positively regulating the progenitor differentiation. It presents as an early marker and should be positive throughout the differentiation process. Collagen II is essential for the normal embryonic development of the skeleton, linear growth, and the ability of cartilage to resist compressive forces. Therefore, COLII would be positive at the late phase of differentiation when cartilage tissue is formed. In our experiment, hASCs were stained with SOX9 after 14 days and with COLII after 21 days of differentiation. Fig. 6 shows the results of immunofluorescence staining. SOX9 and COLII were labeled with green fluorescence. For SOX9, all four groups showed a positive signal since most cells were positively stained; this proved a differentiated trend. However, the D150 groups showed an integrated positive signal that included the entire cytoplasm of the hASCs. This might suggest a high differentiation ratio of hASCs in the D150 group. For COLII staining, a different result was observed. In the D60 group, no positive signal was observed.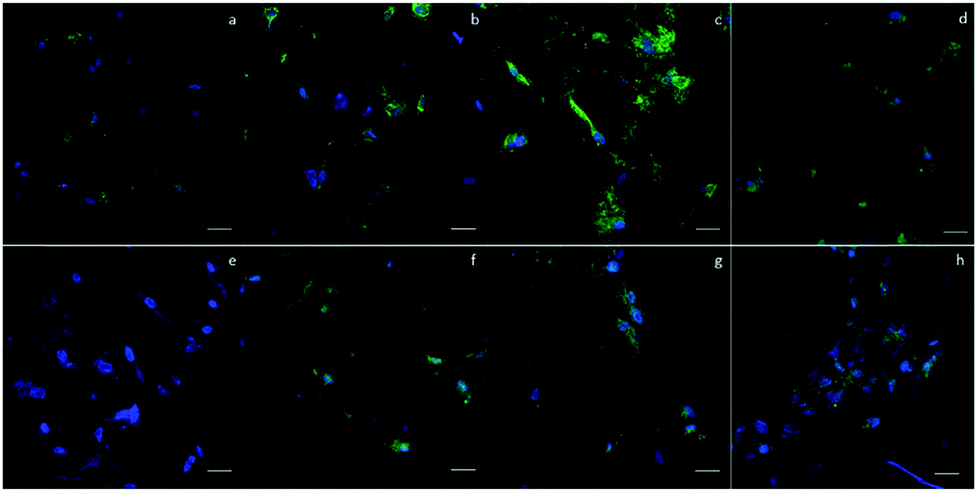 | ||
| Fig. 6 Immunofluorescence staining of (a)–(d) SOX9 in four groups of scaffolds after 14 days of differentiation. (e)–(h) COLII in four groups of scaffolds after 21 days of differentiation. Fewer positive signals were observed in smaller pore-size (a) and (e) D60 groups. Highly positive results were observed in larger pore size (c) and (g) D150 and (d) and (h) D190 groups. Green signal indicates the targeted antigen SOX9 or COLII. Blue signal indicates nucleus. Scale bar = 100 μm. | ||
In the other groups, a positive COLII signal was obtained. This result possibly indicates that a small pore size may not be able to induce full chondrogenesis, explaining the positive signal of SOX9 and negative signal of COLII.26,27
3.5 Glycosaminoglycan (GAG) quantification
After a positive signal was observed with immunofluorescence staining, we wanted to verify chondrogenic differentiation through secretion of extra cellular matrix (ECM). Cartilage tissues are enriched in glycosaminoglycans (GAGs). Therefore, GAG should be detected if hASCs go through mature chondrogenic differentiation in the scaffolds. To examine the GAG content in our scaffolds, we first conducted the compression test, as abovementioned in previous sections. If GAGs were secreted, the stiffness of the scaffolds should increase.28 To remove the possible influence of cells towards measured stiffness, a scaffold plus cells group was added in the experiment simply by conducting a compression test 1 day after seeding. As shown in Fig. 7(a), in zone I, the elasticity of the scaffolds increased after seeding in the D60 and D100 groups, but there was no increase in the differentiated groups. For D150 and D190 groups, the elasticity of the scaffold simply increased as cells were seeded and cultured. However, there was no obvious trend in zone I. In zone III, Fig. 7(b) showed that there was no significant difference in stiffness regardless of whether the cells were seeded or not. Thus, in most groups, an increase in stiffness was observed after differentiation for 21 days. This indicated that the contents of the scaffold and extracellular matrix were changed. This change led to an increase in stiffness of the scaffold in all the groups. As a result, there should be extracellular matrix secretion during differentiation, and further experiments were conducted to ensure that GAG was secreted.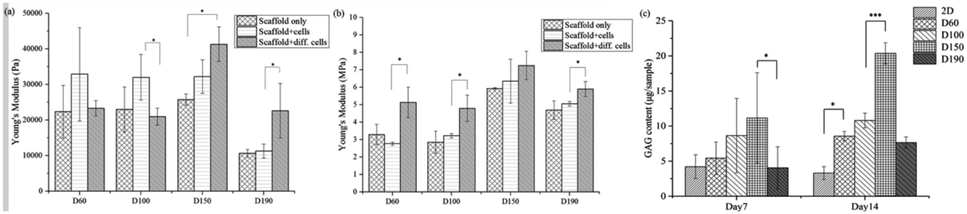 | ||
| Fig. 7 Young's modulus of the scaffold, scaffold with cell cultured for 1 day, and scaffold with hASCs under differentiation for 21 days from the stress–strain curve obtained during the compression test in (a) zone I and (b) zone III. (c) GAG contents of the scaffolds that were seeded with hASC and differentiated for 7 and 14 days. | ||
The GAG content was measured via dimethylmethylene blue (DMMB) assay. Since the DMMB assay quantifies the GAG content of the sample with a calibration curve, it would be a more accurate way to calculate the GAG content. Fig. 7(c) shows the GAG content in every scaffold after 7 and 14 days of chondrogenic differentiation.29 At day 7, the 2D and D190 groups contained the lowest GAG content, whereas the D150 group showed the highest GAG content. Aside from the D190 groups, the other groups showed a trend that as the pore size increased, the GAG content increased. The D190 groups did not fit with the trend possibly due to the relatively low elasticity. However, we could see that the GAG content increased in all the 3D groups as the differentiation time was prolonged from 7 days to 14 days. Moreover, to ensure that there were similar amounts of cells in each scaffold, we referred to the results of MTT (Fig. 4(f)). Both results showed that when the seeding density was the same in each scaffold, the proliferation rates were around the same level after a period of time. This indicated that the cell number should be similar in all four groups while conducting the DMMB assay. As a result, this confirmed GAG secretion by hASCs during chondrogenic differentiation and was an important benchmark for differentiation.
3.6 Gene expression
To evaluate the specific chondrogenic marker expression of hASCs after differentiation, qPCR was used as the most convenient technique. There were four gene markers used: SOX9, AGG, COLII, and COLI. SOX9 is the main transcription factor for chondrogenesis that expresses in every stage of differentiation. AGG is a marker that expresses at the late stage of differentiation. During the formation of the cartilage matrix, the AGG marker is upregulated and promotes the deposition of the cartilage-specific ECM. COLI is a marker that would be positive in the pre-cartilage matrix stage and negative after the mature cartilage tissue is formed.30 Therefore, SOX9 and AGG were used as positive markers, whereas COLI was used a negative marker. COLII is present at the late phase of differentiation when cartilage tissue is formed. As a result, for only 7 and 14 days of culturing, the gene expression of COLII is not significantly different in each group. Thus, we only showed the gene expression for differentiation for 21 days. Fig. 8(a) and (b) show the qPCR result of SOX9 and AGG after 7 and 14 days of differentiation. By comparing the expression at different time points, we could see that both SOX9 and AGG were relatively low at day 7. However, at day 14, there was a significant increase in SOX9 and AGG expression. Moreover, both gene expressions were significantly higher in the D150 groups. D60 showed relatively low expression of SOX9 and AGG; this corresponded to the results of the GAG content and immunofluorescence staining. Therefore, there was a trend that as the pore size increased, there was an advantage towards hASC chondrogenic differentiation. In addition, 2D control groups were extremely low in expression as compared to the 3D groups. To ensure the credibility of the 2D groups, a COLI expression test was conducted to examine whether the 2D groups lacked the ability to induce or maintain chondrogenic phenotype. Fig. 8(c) and (d) show that the COLI level in the 2D control increases significantly from day 7 to day 21. As a negative control, all 3D groups sustained around the same level of COLI as the differentiation days prolonged. These results showed that hASC differentiation potential was highly affected by the pore size.31 Moreover, this further confirmed chondrogenic differentiation in 3D groups since hASCs without differentiation showed a high level of COLI through cell culture. | ||
| Fig. 8 Gene expression of (a) SOX9 (b) AGG, and (c) COLI after 7, 14, and 21 days of differentiation. COLI was examined as a negative control primer. (d) The amplified version of day 21 expression of COLII from (c). *p < 0.05. **p < 0.01. ***p < 0.001. | ||
4. Conclusions
Bubble size, or pore size, is a crucial factor in cell behavior and differentiation studies. In this study, we fabricated four non-cytotoxic gelatin scaffolds with homogeneous pores but different pore sizes. Elasticity and stiffness were also measured to ensure that all four groups had the same level of mechanical properties.hASC morphology was significantly influenced by the pore size. This led to the following results of chondrogenic differentiation. hASCs in D150 showed highly positive signals in immunofluorescence staining of SOX9 and COLII. Moreover, they showed high GAG content after 14 days of differentiation. In qPCR, D150 showed high gene expression of AGG, SOX9, and COLII, whereas COLI was significantly low in all 3D groups as compared to the case of the 2D control; this further confirmed the differentiation of hASCs in 3D scaffolds. In conclusion, D150, which is the scaffold with the largest pore size, shows the highest chondrogenic differentiation potential of hASCs.
In this study, we investigated the influence of pore size on chondrogenic differentiation. To induce mature chondrogenesis, a large pore size is required. This study has confirmed the abovementioned statement, and further applications can be investigated based on this result.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The hASCs were obtained from the National Taiwan University, Department of Surgery. This work was supported by the Ministry of Science and Technology, Taiwan (MOST 104-2221-E-002-124-MY3).References
- S. B. Lee, Y. H. Kim, M. S. Chong, S. H. Hong and Y. M. Lee, Biomaterials, 2005, 26, 1961–1968 CrossRef CAS PubMed.
- M. Brittberg, A. Lindahl, A. Nilsson, C. Ohlsson, O. Isaksson and L. Peterson, N. Engl. J. Med., 1994, 331, 889–895 CrossRef CAS PubMed.
- A. Schäffler and C. Büchler, Stem Cells, 2007, 25, 818–827 CrossRef PubMed.
- P. A. Zuk, M. Zhu, P. Ashjian, D. A. De Ugarte, J. I. Huang, H. Mizuno, Z. C. Alfonso, J. K. Fraser, P. Benhaim and M. H. Hedrick, Mol. Biol. Cell, 2002, 13, 4279–4295 CrossRef CAS PubMed.
- B. P. Mahadik, T. D. Wheeler, L. J. Skertich, P. J. Kenis and B. A. Harley, Adv. Healthcare Mater., 2014, 3, 449–458 CrossRef CAS PubMed.
- S. A. Abdellatef, A. Ohi, T. Nabatame and A. Taniguchi, Int. J. Mol. Sci., 2014, 15, 4299–4317 CrossRef PubMed.
- A. Higuchi, Q.-D. Ling, Y. Chang, S.-T. Hsu and A. Umezawa, Chem. Rev., 2013, 113, 3297–3328 CrossRef CAS PubMed.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- W. Chen, L. G. Villa-Diaz, Y. Sun, S. Weng, J. K. Kim, R. H. Lam, L. Han, R. Fan, P. H. Krebsbach and J. Fu, ACS Nano, 2012, 6, 4094–4103 CrossRef CAS PubMed.
- M. Witkowska-Zimny, K. Walenko, A. E. Wałkiewicz, Z. Pojda, J. Przybylski and M. Lewandowska-Szumieł, Acta Biochim. Pol., 2012, 59, 15–24 Search PubMed.
- A. Marino, G. Ciofani, C. Filippeschi, M. Pellegrino, M. Pellegrini, P. Orsini, M. Pasqualetti, V. Mattoli and B. Mazzolai, ACS Appl. Mater. Interfaces, 2013, 5, 13012–13021 CAS.
- J. M. Szymanski and A. W. Feinberg, Biofabrication, 2013, 6, 024104 CrossRef PubMed.
- A. A. Jaeger, C. K. Das, N. Y. Morgan, R. H. Pursley, P. G. McQueen, M. D. Hall, T. J. Pohida and M. M. Gottesman, Biomaterials, 2013, 34, 8301–8313 CrossRef CAS PubMed.
- Z.-Z. Zhang, D. Jiang, J.-X. Ding, S.-J. Wang, L. Zhang, J.-Y. Zhang, Y.-S. Qi, X.-S. Chen and J.-K. Yu, Acta Biomater., 2016, 43, 314–326 CrossRef CAS PubMed.
- B. Sun, X.-J. Jiang, S. Zhang, J.-C. Zhang, Y.-F. Li, Q.-Z. You and Y.-Z. Long, J. Mater. Chem. B, 2015, 3, 5389–5410 RSC.
- H. Shen, Y. Niu, X. Hu, F. Yang, S. Wang and D. Wu, J. Mater. Chem. B, 2015, 3, 4417–4425 RSC.
- C. A. Vissers, J. N. Harvestine and J. K. Leach, J. Mater. Chem. B, 2015, 3, 8650–8658 RSC.
- H.-W. Kang, Y. Tabata and Y. Ikada, Biomaterials, 1999, 20, 1339–1344 CrossRef CAS PubMed.
- Y. Hiraki, C. Shukunami, K. Iyama and H. Mizuta, Osteoarthritis and Cartilage, 2001, 9, S102–S108 CrossRef PubMed.
- C. Merceron, C. Vinatier, S. Portron, M. Masson, J. Amiaud, L. Guigand, Y. Chérel, P. Weiss and J. Guicheux, Am. J. Physiol.: Cell Physiol., 2010, 298, C355–C364 CrossRef CAS PubMed.
- E. M. Weijers, L. J. Van Den Broek, T. Waaijman, V. W. Van Hinsbergh, S. Gibbs and P. Koolwijk, Tissue Eng., Part A, 2011, 17, 2675–2685 CrossRef CAS PubMed.
- B. T. Estes, B. O. Diekman, J. M. Gimble and F. Guilak, Nat. Protoc., 2010, 5, 1294 CrossRef CAS PubMed.
- P. Angele, R. Müller, D. Schumann, C. Englert, J. Zellner, B. Johnstone, J. Yoo, J. Hammer, J. Fierlbeck and M. K. Angele, J. Biomed. Mater. Res., Part A, 2009, 91, 416–427 CrossRef PubMed.
- J. Garcia, C. Mennan, H. S. McCarthy, S. Roberts, J. B. Richardson and K. T. Wright, Stem Cells Int., 2016, 2016, 195–200 Search PubMed.
- S. H. Oh, T. H. Kim, G. I. Im and J. H. Lee, Biomacromolecules, 2010, 11, 1948–1955 CrossRef CAS PubMed.
- L. Reppel, J. Schiavi, N. Charif, L. Leger, H. Yu, A. Pinzano, C. Henrionnet, J.-F. Stoltz, D. Bensoussan and C. Huselstein, Stem Cell Res. Ther., 2015, 6, 260 CrossRef PubMed.
- S. V. Naveen, R. E. Ahmad, W. J. Hui, A. M. Suhaeb, M. R. Murali, R. Shanmugam and T. Kamarul, Int. J. Med. Sci., 2014, 11, 97 CrossRef PubMed.
- C. C. Wang, K. C. Yang, K. H. Lin, C. C. Wu, Y. L. Liu, F. H. Lin and I. H. Chen, Biotechnol. Bioeng., 2014, 111, 2338–2348 Search PubMed.
- P. Singh and J. E. Schwarzbauer, J. Cell Sci., 2012, 125, 3703–3712 CrossRef CAS PubMed.
- Y.-P. Lo, Y.-S. Liu, M. G. Rimando, J. H.-C. Ho, K.-h. Lin and O. K. Lee, Sci. Rep., 2016, 6, 21253 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tb02244a |
| ‡ K. H. Wu and C. Mei contribute equally to this study. |
| This journal is © The Royal Society of Chemistry 2018 |