
Co-delivery of PLK1-specific shRNA and doxorubicin via core-crosslinked pH-sensitive and redox ultra-sensitive micelles for glioma therapy†
Pu
Wang‡
a,
Nengwei
Yu‡
b,
Yi
Wang‡
a,
Huili
Sun
a,
Zhenglin
Yang
b and
Shaobing
Zhou
 *a
*a
aKey Laboratory of Advanced Technologies of Materials, Ministry of Education, School of Materials Science and Engineering, Southwest Jiaotong University, Chengdu, P. R. China. E-mail: shaobingzhou@swjtu.edu.cn; shaobingzhou@hotmail.com; Tel: +86-28-87634068
bDepartment of Neurology, Sichuan Academy of Medical Sciences and Sichuan Provincial People's Hospital, Chengdu, Sichuan, China
First published on 29th November 2017
Abstract
Anticancer drug delivery encounters many biological barriers, including mucosal barriers, nonspecific uptake and intracellular drug resistance. Consequently, efficient delivery of therapeutic agents with nanocarriers to the target cell and controlled intracellular release of encapsulated drugs are key to achieving high therapeutic efficiency. In this study, we develop a tumor microenvironment-sensitive polymer micelle system from a pH- and glutathione (GSH) dual-responsive copolymer with each repeating unit containing a disulfide bond. To prevent premature drug release within the blood circulation, the core region was chemically crosslinked via UV light irradiation. In tumor cells, the micelles are able to escape from the acidic lysosome into the cytoplasm via a prompt expansion due to the “proton sponge effect”. Subsequently, ultra-sensitive redox responsiveness is realized since the abundant disulfide bonds of the micellar matrix can be cleaved by a high level of GSH, leading to a rapid intracellular release of encapsulated doxorubicin (DOX) and PLK1-specific shRNA. The antitumor activity in U87 glioma tumor-bearing mice reveals that this novel system possesses a high therapeutic efficacy against solid tumors with negligible side effects on normal tissues. Therefore, this micellar nanoplatform has great potential in delivering drugs for enhanced glioma therapy.
1 Introduction
Nanocarriers are considered to be a promising tool in the delivery of drugs and genes.1,2 Based on their well-known enhanced permeation and retention (EPR) effect,2,3 nanocarriers can deliver encapsulated drugs and genes preferentially into cancer tissues, significantly enhance the therapeutic efficacy or transfection efficacy and reduce the side effects of their cargo.4 Polymeric micelles are one of the well-studied nanocarriers that have a unique architecture containing a hydrophobic core and hydrophilic outer shell, and they are formed by self-assembly of amphiphilic copolymers.5,6 In comparison with other nanocarriers, they have more advantages, such as an enhancement of the solubility of drugs with low water solubility, an extension of in vivo blood circulation due to their ability to shield the drug from the reticular endothelin system (RES), and resistance to dilution due to their low critical micelle concentration (CMC).7,8 The advantages of these micellar nanocarriers have greatly improved the delivery of drugs and genes to their target sites; however, the practical drug accumulation efficiency and anticancer effect are still lower than expected.9 One of the reasons may be that the micellar system is not very stable in the blood circulation, resulting in a leakage of its cargo at unexpected sites.2 On the other hand, even if the drug-loaded micelles are internalized by the cancer cells, they would still encounter other obstacles such as early endosomes, late endosomes, and lysosomes, which make it challenging to achieve effective intracellular drug release.In this study we developed a core-crosslinked polymeric micelle nanoplatform with pH/redox dual-responsivity. The crosslinked core of the micelle can prevent leakage of the encapsulated drugs within the blood circulation.10 Simultaneously, the stimuli-responsive functions can achieve a triggered drug release to the target sites in the tumor cells.11–13 Among these stimuli-responsive drug delivery systems, the pH-responsive nanocarrier is one of the most well-studied nanocarriers because pH values vary in different tissues and cellular compartments,14–16 and this can help to achieve pH-triggered drug release.17–19 In addition, redox stimulation is another well-known potent stimulus of nanocarriers containing disulfide linkages for controlled intracellular drug release, because the excess glutathione (GSH) in the cytoplasm of cancer cells can effectively break the disulfide bonds.11,20–22 However, for previous redox-responsive micelles, the disulfide linkages were generally introduced between the hydrophobic and hydrophilic segments of the amphiphilic block copolymers.23–25 Although these redox responsive micelles can respond to the intracellular GSH with relatively high concentrations through the cleavage of the hydrophobic and hydrophilic segments of the copolymers, the residual hydrophobic segments are still macromolecules with long chains, which become a barrier for intracellular drug release. To address this limitation, in this work we designed a novel amphiphilic block copolymer, in which the disulfide bonds were introduced to form the backbone of the polymer chain. Unlike other redox-responsive polymer micelles with only one disulfide bond in each polymer chain, this copolymer contains many disulfide bonds in a single polymer chain since each repeating unit of the polymer backbone contains a disulfide bond. Thus, the micelles have an ultra-sensitive redox function. In addition, to enhance the anticancer effect, a frequently used strategy is to co-deliver a dual anticancer drug in combination with the nanocarriers.26
Because 3-(dibutylamino) propylamine (DBP) is hydrophobic in neutral environments, it facilitates DOX loading, and at the same time, it can be converted into hydrophilic under acidic conditions, which can greatly promote the release of DOX when the micelles are internalized by cancer cells.27 We synthesized a polymer based on acrylate-amine Michael Addition with DBP, PEG and N,N′-cystaminebisacrylamide. In addition, the polymer has a higher positive charge and can transport RNA. Accordingly, in this study, we developed a pH/redox dual-responsive core-crosslinked micellar platform to co-deliver a polo-like kinase 1 (PLK1) specific shRNA and DOX for enhanced chemotherapy of glioma tumors (Scheme 1a). Dual-drug delivery systems have attracted more and more attention due to the fact that combined therapy with drugs of different therapeutic effects is an effective method in the treatment of diseases and tissue repair, maximizing therapeutic efficacy, overcoming drug resistance and diminishing side effects.28–30 In particular, in cancer therapy, combinations of multiple drugs have been successfully developed to overcome the drug resistance in cancer therapy.31 Dual-/multiple-drug loaded stimuli-responsive micelles can further enhance the anticancer effect. For example Chen et al.32 have reported dual-drug-loaded smart micelles based on a mixture of poly(propylene oxide)-b-poly(γ-benzyl-L-glutamate)-b-poly(ethylene glycol) (PPO-b-PBLG-b-PEG) triblock terpolymers and two model drugs, doxorubicin (DOX) and naproxen (Nap), where the DOX is chemically linked to the PBLG backbones through an acid-cleavable hydrazone bond, and the Nap is physically encapsulated in the cores. These micelles are biocompatible under normal physiological conditions and possess a maximized therapeutic effect against cancer cells.
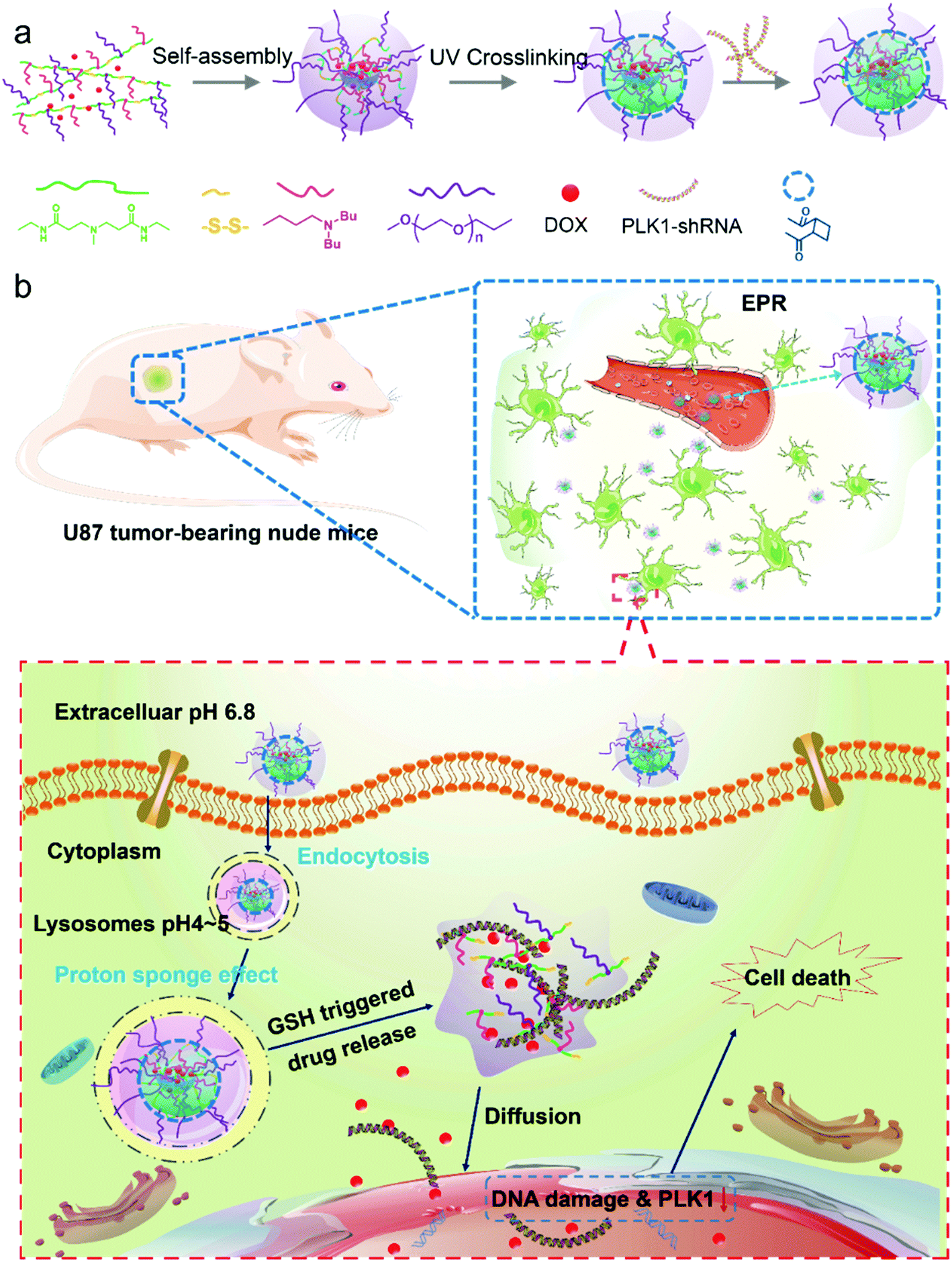 | ||
| Scheme 1 (a) A schematic illustration of the self-assembly and UV crosslinking process of the dual-responsive micelles. (b) A schematic illustration of a glioma and co-delivery of shRNA and doxorubicin into a glioma cell via core-crosslinked pH- and redox-sensitive micelles based on the EPR effect. | ||
In our study, the designed micelles can be accumulated in the tumor tissue via the enhanced penetration and retention (EPR) effect; and subsequently be internalized into tumor cells by endocytosis (Scheme 1b). The micelles will escape quickly from the acidic lysosomes due to the pH triggered proton sponge effect, and once in the cytoplasm they will be “blasted” into small molecules (like a bomb) in response to the intracellular GSH with a high concentration, leading to a rapid release of the encapsulated drugs. Finally, the DOX and shRNA will diffuse into the nucleus (Scheme 1b). As a result, DOX molecules will integrate with nuclear DNA, causing DNA damage, and the PLK1-specific shRNA will down-regulate the expression of PLK1, which is related to the formation of centrosomes, chromosomes and tumor development. Dysregulation of PLK1 in normal cells has been found to result in the formation of abnormal centrosomes, a transformed phenotype and increased tumorigenicity.33,34 As a result, the co-delivery of PLK1-specific shRNA and DOX can enhance the anticancer effect of the drug-loaded micelles. In addition, a glioma tumor model, which is a model of one of the most poorly treated cancers,35 is employed.
2 Experimental section
2.1 Materials
Acrylic acid, cysteamine hydrochloride, monomethyl poly(ethylene glycol) (mPEG, MW = 2000), glutathione (GSH) and 4,6-diamidino-2-phenylindole (DAPI) were purchased from Sigma-Aldrich (USA). Polo-like kinase 1 specific Short hairpin RNA (PLK1-shRNA) was purchased from Shanghai Sangon Biotech Co., Ltd. Doxorubicin hydrochloride (DOX·HCl) was purchased from Zhejiang Hisun Pharmaceutical Co., Ltd (China). Annexin V-FITC Apoptosis Detection Kits were obtained from KeyGEN Biotech (China). All the other chemical reagents were purchased from commercial suppliers and used without further purification.2.2 Cells and animals
Glioblastoma cells (U87 and U251) and endothelial cells (ECs) were obtained from Cell Bank of Chinese Academy of Sciences and Sichuan University (China), respectively. Cells were cultured in RPMI 1640 with fetal bovine serum (FBS, 10%, v/v) at 37 °C and 5% CO2 under fully humidified conditions.Female nude mice (20 ± 2 g) at 5–6 weeks of age, were purchased from the Experimental Animal Center of Sichuan University and fed under conditions of 25 °C and 55% humidity. Animal experiments were approved by the Institutional Animal Care and Use Committee of Sichuan Academy of Medical Sciences and Sichuan Provincial People's Hospital (P. R. China), and all protocols for this animal study conformed to the Guide for the Care and Use of Laboratory Animals.
2.3 Synthesis and characterization of copolymers
2.4 Fabrication and characterization of micelles
The DOX-loaded micelles or DOX/shRNA-loaded micelles were prepared using similar procedures, except that a certain amount of DOX or shRNA was dissolved in the THF together with the different copolymers. In addition, all processes were carried out in the dark except for the UV irradiation for 15 min. Then, the final drug-loaded micelle solution was transferred into a dialysis bag (MWCO 1000) with deionized water to remove the free drugs. All the micelle solution samples were lyophilized and stored at −20 °C.
2.5 GSH-triggered or pH-triggered disassembly behaviors of the micelles
To evaluate the GSH-triggered or pH-triggered disassembly behaviors, changes in micellar size distribution in response to phosphate buffered saline (pH 7.4) containing GSH (10 mM) or acetate buffered solution (ABS) at pH 5.0, were investigated by DLS measurements. The morphology changes in the micelles before and after responding to the dual stimulations were measured using TEM.2.6 In vitro drug release study
The in vitro release profiles of DOX from the dual responsive micelles in different release media, were investigated, including: PBS (pH 7.4) with 10 μM or 10 mM GSH and ABS (pH 5.0) with 10 μM or 10 mM GSH. The freeze-dried micelle sample was dissolved in water (1 mg mL−1) and 1.0 mL of this solution was transferred into a dialysis bag (MWCO 1000), followed by immersion of the dialysis bag into a tube containing 30 mL of incubation medium. These samples were kept in a shaking bed at 100 rpm and 37 °C. The amount of released drug in the incubation medium was quantified by a fluorescence spectrophotometer (F-7000, Hitachi, Japan).2.7 Cytocompatibility evaluation
The cytocompatibility of the blank micelles was evaluated by an Alamar blue assay with glioblastoma cells (U87 and U251) and EC cells. Cells were seeded in a 48-well plate at a density of 1 × 104 cells per well and cultured for 24 h. Then, the blank micelle solution was added with concentrations ranging from 10 μg mL−1 to 200 μg mL−1 for another 24 h of incubation. After that, the medium was removed and the cells were washed three times with PBS, followed by the addition of 300 μL of Alamar blue solution (10% Alamar blue, 10% FBS and 80% media 199 (Gibco), v/v) and a 3 h incubation. The Alamar blue solution was then transferred into a 96-well plate and read by a microplate spectrophotometer (ELX800 Biotek, USA) at 570 nm (excitation)/600 nm (emission). Finally, live cell staining was performed (cells were stained with 1 μM calcein AM [Sigma, USA]) and the cells were observed by fluorescence microscopy (CKX41, Olympus, Japan).2.8 Intracellular drug release and in vitro gene transfection
The cellular internalization and intracellular release behaviors of the in vitro gene transfection of the DOX/shRNA-micelles, shRNA-micelles and shRNA-PEI were investigated by confocal laser scanning microscopy (CLSM, FV1000, Olympus, Japan) and flow cytometry (FCM, BD Accuri C6, USA) with U87 cells. After incubation for 1 h or 5 h, the cells were then fixed with 2.5 wt% glutaraldehyde for 50 min and stained with DAPI for 10 min. Then, they were observed by CLSM. For flow cytometric analysis, after pre-setting the incubation time, cells were washed three times with PBS and retrieved by treatment with trypsin and centrifugation at 2000 rpm for 3 min. Next, flow cytometry was used to analyze the collected cells.2.9 In vitro antitumor activity assay
The anticancer activity of the free DOX, DOX-loaded micelles (DOX-micelles), DOX/shRNA-loaded micelles (DOX/shRNA-micelles), shRNA-loaded micelles (shRNA-micelles) and shRNA-loaded PEI nanoparticles (shRNA-PEI) was evaluated by Alamar blue assay with U87 cells. Briefly, U87 cells were seeded in a 48-well plate at a density of 1 × 104 cells per well. After 24 h, the samples were added to incubate for another 24 h. The DOX dose ranged from 0.0625 μg mL−1 to 8 μg mL−1. The shRNA dose ranged from 0.005 μg mL−1 to 1 μg mL−1. The procedures for the Alamar blue assay were similar to those described above.2.10 Cell apoptosis assay
To confirm the anticancer effect of the dual responsive DOX or shRNA-loaded micelles, a cell apoptosis assay was performed by FCM. Briefly, U87 cells were seeded in 6-well plates at a density of 1 × 105 cells per well and incubated for 24 h. Then, the cells were treated with different samples as mentioned in Section 2.9 and incubated for another 24 h. The DOX dose was 4 μg mL−1 and the shRNA dose was 0.4 μg mL−1. The Annexin V-FITC Apoptosis Detection Kit was used to determine the apoptosis rate according to the manufacturer's protocol.2.11 Tumor modeling and in vivo DOX biodistribution
To establish tumor models, 100 μL of a cell suspension containing 1 × 106 U87 cells was inoculated subcutaneously in the right flank of the nude mice. The tumor volume was measured with a Vernier caliper across its longest (a) and shortest (b) diameters and calculated with the equation, volume = ab2/2. Approximately 10 days post inoculation, the tumors in the mice reached a volume of approximately 25 mm3 (labeled as day 0).For evaluation of the in vivo micelle distribution, U87 tumor-bearing nude mice were administered saline (in the control group), and free DOX and DOX-micelles via the lateral tail vein at a 5 mg kg−1 equivalent DOX dose. At 1, 3, and 6 h post administration, the mice were anesthetized and imaged with a Maestro in vivo imaging system. At 6 h, organs and tumors from the sacrificed mice were also imaged.
2.12 In vivo antitumor study
To evaluate the in vivo antitumor effect of the different samples, U87 tumor-bearing nude mice were randomly divided into five groups (n = 7) and received intravenous administration of blank micelles, free DOX, DOX-micelles, shRNA-micelles and DOX/shRNA-micelles at 5 mg kg−1 equivalent DOX and 0.5 mg kg−1 equivalent shRNA, respectively. The intravenous administration was performed every two days, 4 times in all. Mouse weight and tumor size were recorded every two days starting at day 0.At day 14, some of the mice from each group were picked randomly and sacrificed to obtain their tumor tissues. Tumor sections of 5 μm were prepared and stained with hematoxylin/eosin (H&E) or terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) for microscopic observation. The mean optical density (OD) values were measured on three pictures from each sample with Image-Pro Plus 6.0 software.
Furthermore, a western blotting analysis and reverse transcription-polymerase chain reaction (RT-PCR) were performed to determine the mechanism of tumor suppression in vivo. For the western blotting analysis, the tumors were lysed at 0 °C by RIPA buffer (RIPA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cocktail = 100:1), centrifuged and the supernatant was collected. The total protein was quantified by the BCA protein assay kit (KGPBCA, KeyGEN). A 100 μg sample of the protein was electrophoresed in 10% SDS-PAGE. Then, it was transferred to a nitro-cellulose membrane, blocked and incubated overnight with monoclonal antibodies (ABCAM). After washing three times with TBST buffer, 2 h of incubation with horseradish peroxidase conjugated secondary antibody (ABCAM), was applied to the membrane, and followed by rinsing three times with TBST buffer. The target protein bands were visualized using the ECL kit (Thermo Scientific). For quantitative RT-PCR, the polo-like kinase 1 (PLK1), proapoptotic gene caspase-3 and antiapoptotic gene BCL2 expression levels from the U87 tumors were determined. Tumors were taken from the mice at day 14 and were homogenized for RNA extraction. The total RNA was isolated from the tumors using TRIzol (Invitrogen, Carlsbad, CA, USA). Then, the RNA was converted to cDNA by the PrimeScript RT reagent Kit (Takara, Dalian, China) according to the manufacturer's protocols. Quantitative real-time PCR was employed to analyze the obtained cDNA samples with an SYBR Premix Ex Taq II Kit (Takara, Dalian, China). The relative levels of mRNA expression were normalized to GADPH expression for each gene and were compared to the saline group. The sequences of the primers for these genes are listed below:
:Cocktail = 100:1), centrifuged and the supernatant was collected. The total protein was quantified by the BCA protein assay kit (KGPBCA, KeyGEN). A 100 μg sample of the protein was electrophoresed in 10% SDS-PAGE. Then, it was transferred to a nitro-cellulose membrane, blocked and incubated overnight with monoclonal antibodies (ABCAM). After washing three times with TBST buffer, 2 h of incubation with horseradish peroxidase conjugated secondary antibody (ABCAM), was applied to the membrane, and followed by rinsing three times with TBST buffer. The target protein bands were visualized using the ECL kit (Thermo Scientific). For quantitative RT-PCR, the polo-like kinase 1 (PLK1), proapoptotic gene caspase-3 and antiapoptotic gene BCL2 expression levels from the U87 tumors were determined. Tumors were taken from the mice at day 14 and were homogenized for RNA extraction. The total RNA was isolated from the tumors using TRIzol (Invitrogen, Carlsbad, CA, USA). Then, the RNA was converted to cDNA by the PrimeScript RT reagent Kit (Takara, Dalian, China) according to the manufacturer's protocols. Quantitative real-time PCR was employed to analyze the obtained cDNA samples with an SYBR Premix Ex Taq II Kit (Takara, Dalian, China). The relative levels of mRNA expression were normalized to GADPH expression for each gene and were compared to the saline group. The sequences of the primers for these genes are listed below:
PLK1 forward: ACCAGCACGTCGTAGGATTC
PLK1 reverse: CAAGCACAATTTGCCGTAGG
GADPH forward: AACGACCCCTTATTGAC
GADPH reverse: TCCACGACATACTCTGCAC
2.13 Statistical analysis
Data are shown as the mean value ± standard deviation. Single factorial analysis of variance (ANOVA) was performed to determine the statistical significance of the data.3 Results and discussion
3.1 Preparation and characterization of the dual responsive copolymer
The multifunctional copolymer, AC–Cys(DBP)–Cys(mPEG)–AC, was synthesized using a “one-pot method”, where a Michael addition occurred through blending the synthesized N,N′-cystaminebisacrylamide (AC–Cys–AC) monomer, 3-(dibutylamino) propylamine (DBP) and mPEG–NH2, as shown in Fig. S1 (ESI†). In this copolymer, tertiary amine groups and the disulfide bonds were simultaneously introduced to form the backbone of the polymer chain by using 3-(dibutylamino) propylamine and N,N′-cystaminebisacrylamide as the Michael addition substrates. In this way, both the disulfide bonds and the acid-sensitive tertiary amines acted as repeating units in the main chains of the polymer. Different from other redox-responsive polymers, each macromolecular chain of this polymer has a large amount of disulfide bonds, ensuring redox ultra-sensitivity. The large number of tertiary amine repeating units endow the micelles with the ability to carry genes and produce pH responsiveness for the proton sponge effect in the lysosome. 1HNMR spectral analysis (Fig. S2 in the ESI†) was also performed to confirm the composition and structures of the resultant polymers. In addition, the double bonds in the terminal of the copolymer can be crosslinked by UV irradiation, which is thought to make a contribution to prepare a shrunken and stable core of the nanocarriers in the followed experiments. The polymer molecular weight as Mn (NMR) in total was 5.47 kDa, and the average repeating numbers of CYS-DBP and CYS-PEG were 1.43 and 5.07 respectively.3.2 Self-assembly and triggered-disassembly of micelles
After successful synthesis of the multifunctional copolymers AC–Cys(DBP)–Cys(mPEG)–AC, they were used to fabricate micelles using a solvent evaporation method. A series of DLS measurements were performed to determine the formation, size distribution, stability and sensitivity to stimulation (an acidic or reductive environment) for the micelles self-assembled from the AC–Cys(DBP)–Cys(mPEG)–AC copolymer, as shown in Fig. 1. Fig. 1a shows the size distributions of the micelles before and after UV irradiation, in which one single peak can be found under both conditions, suggesting that whether the micelles are crosslinked or not, they have a uniform size distribution. Furthermore, after the UV irradiation, the average diameter of the micelles changed from 44 nm to 73 nm, which may be proof of the successful crosslinking and shrinkage of the micellar core because only the double bonds in the terminal of the hydrophobic segments of the copolymer can be crosslinked by UV irradiation. The 1H NMR spectra of the UV-crosslinked micelles and the non-crosslinked micelles are shown in Fig. S3 (ESI†). From Fig. S3 (ESI†), the characteristic peaks of acryloyl appeared in the polymers without UV-crosslinking, and after UV-crosslinking, the characteristic peaks of acryloyl disappeared which confirmed that the micelles were crosslinked by UV. | ||
| Fig. 1 Characterization of polymeric micelles. (a) Size distribution of crosslinked or non-crosslinked micelles. (b) Average size changes of the crosslinked micelles over time. (c) Change in size distribution profiles of the crosslinked double responsive micelles at pH 5.0 against time. (d) Change in size distribution profiles of the micelles triggered by 10 mM GSH at pH 7.0. (e) Change in size distribution profiles of the micelles in the presence of 10 mM GSH in PBS at pH 5.0. (f) The corresponding TEM images of the micelles before and after the treatment with PBS containing 10 mM GSH at pH 5.0 for 6 h. | ||
The average diameter of the crosslinked micelles remained almost unchanged after 28 days (Fig. 1b), demonstrating that they are stable in an aqueous solution and can be further utilized in cell culture and animal experiments. The zeta potential distribution of the core-crosslinked micelles was also measured as shown in Fig. S4 (ESI†). The average zeta potential of the micelles is ∼28 mV, which helps with the absorption of the gene drugs with a negative charge and stabilizes the dispersion of the micelles.
To demonstrate the pH- and reduction–sensitivity of the micelles, the size distributions of the crosslinked micelles under different stimulant conditions were measured. Fig. 1c shows the size distribution of the cross-linked micelles at pH 5.0 for 1 h. It can be seen that after 30 min of incubation, the core crosslinked micelles display a single peak and the average diameter changed from 44 nm to 73 nm, indicating the swelling of the micelles in response to the acidic environment (pH 5.0), which is mimicking the reducing environment of the lysosome. In order to better understand the micellar size changes in the early endosomes (pH 6.0) and the late lysosomes (pH 5.0), we investigated the size of the micelles under the corresponding pH values, which is shown in Fig. S6 (ESI†). From Fig. S6 (ESI†), it is clearly seen that the size of the micelles in pH 5.0 mimicking late lysosomes is relatively larger than that in pH 6.0 mimicking the early endosomes. This is because there are many unsaturated nitrogen elements contained in the backbone of the polymer chain that attract a large amount of protons and cause the expansion of the micelles, namely, the “proton sponge effect”.39 After 1 h of incubation, multiple peaks can be seen in the size distribution profiles. When 10 mM of GSH was introduced, the size distribution began to change from a narrow single peak to a relatively wide peak after 0.5 h of incubation, and the average size of the micelles increased with the incubation time, reaching 480 nm at 6 h (Fig. 1d), indicating the rapid disassembly of these micelles in an environment that mimics the reducing one of the tumor cells.40 The reason for this is that the backbone of the copolymer contains a large amount of disulfide bonds. Furthermore, the size distribution of the micelles in the presence of both pH 5.0 and 10 mM of GSH was also measured (Fig. 1e). It can be seen that the size distribution shifted from a unimodal peak to irregular multimodal peaks with an increase in incubation time, and after 6 h, no peak can be found, which may be due to the micelles having been expanded and broken into tiny pieces, like a “bomb”, and these pieces dissolved in the solution. To further confirm the triggered destruction of the micelles, TEM observation of the micelles before and after simultaneous stimulation with an acid and a reducer is shown (Fig. 1f). Before stimulation, the micelles displayed as relatively uniform spheres while they changed into indistinguishable small pieces with different sizes, which is confirmed by the DLS measurements. In general, these results suggest that the prepared micelles are pH- and redox-sensitive and can be triggered for destruction, and thus they may be suitable as nanocarriers for intracellular delivery of drugs in cancer therapy.
3.3 In vitro pH- and GSH-triggered drug release
To verify whether or not the micelles are able to rapidly release their cargo under conditions similar to the tumor microenvironment, in vitro pH- and GSH-triggered drug release from the DOX-loaded AC–Cys(DBP)–Cys(mPEG)–AC micelles was investigated. The loading content and encapsulation efficiency of DOX are approximately 6.8% and 65%, respectively. From the DOX release profiles in Fig. 2a, we can see that the cumulative release of DOX in the solution at pH 7.4 with 10 μM of GSH, was less than 20% in 36 h, suggesting that DOX-loaded micelles would be stable in the blood circulation. In contrast, a rapid release of DOX of over 50% in 6 h was triggered immediately with 10 mM of GSH at pH 7.4, which is higher than that of the micelles containing disulfide bond linkages only between the hydrophobic and hydrophilic segments.25,41–43 Similar results were obtained at pH 5.0 with 10 μM of GSH (approximately 43% in 6 h). Moreover, the most rapid DOX release was determined when the dual stimuli (pH 5.0 and 10 mM GSH) were applied simultaneously. These results are consistent with the study of the triggered disassembly of micelles, which was discussed in Section 3.2. Furthermore, this pH- and GSH-triggered release is only expected in cancer treatment, because the pH value of the early endosomes in tumor cells is approximately 5–6, and the late lysosomes are much more acidic at pH 4–5.16,44 Additionally, the reducing environment of the tumor cells (10 mM) also contributes to the DOX release.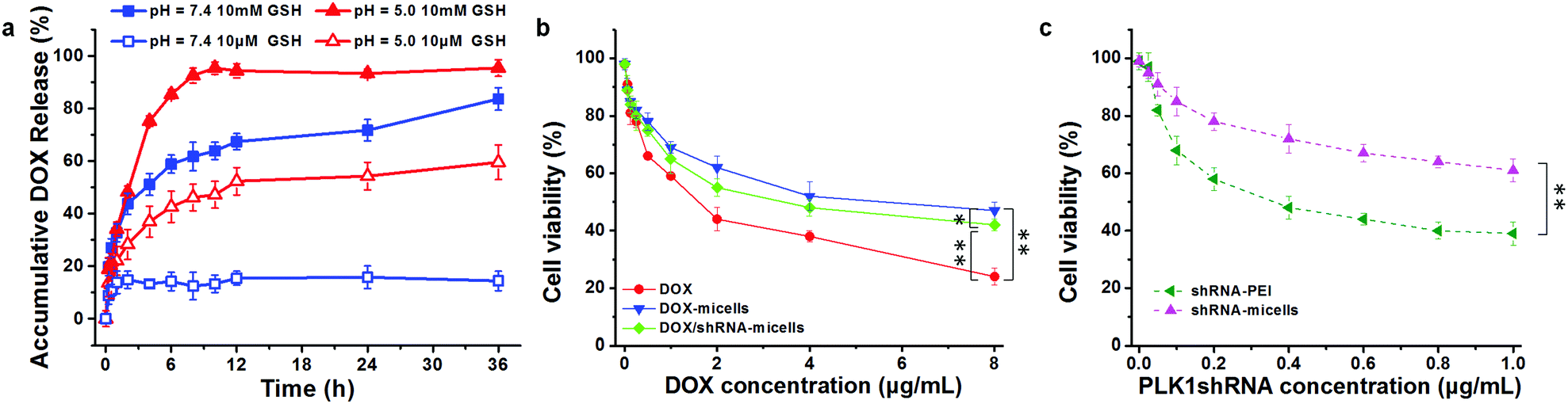 | ||
| Fig. 2 (a) In vitro DOX release from the micelles triggered with GSH (10 μM or 10 mM) in PBS (pH 7.4) or ABS (pH 5.0) at 37 °C. (b) Cytotoxicity of U87 cells treated with free DOX, DOX-loaded micelles and DOX/PLK1 shRNA-loaded micelles as a function of DOX concentration (n = 3). (c) Cytotoxicity of U87 cells treated by PLK1-shRNA-PEI nanoparticles and shRNA-loaded micelles as a function of shRNA concentration (n = 3) (* represents p < 0.05, ** represents p < 0.01). | ||
3.4 Cytocompatibility analysis
Fig. S5 in the ESI† shows the viability and corresponding representative fluorescence images of the EC cells and the glioblastoma cells (U87 and U251) treated with blank AC–Cys(DBP)–Cys(mPEG)–AC micelles. It can be seen that over 90% of the cells remained viable when the concentration of blank micelles ranged from 10 to 250 μg mL−1 (Fig. S5a, ESI†). Additionally, the live cell staining images in Fig. S5b (ESI†) show that all the cell morphologies look normal and healthy. These results indicate that this nanocarrier is cytocompatible and is suitable for further cell and animal experiments.3.5 Intracellular drug release and in vitro gene transfection
To determine the drug release behavior of the DOX-loaded micelles and the transfection effect of shRNA-loaded micelles or PEI nanoparticles in cancer cells, U87 cells were observed by confocal laser scanning microscopy (CLSM) after incubation with DOX, shRNA-micelles, DOX/shRNA-micelles and shRNA-PEI for 1 or 5 h, respectively. For intracellular DOX release, we can see that for all groups, the fluorescence intensity of the cells increased significantly after 5 h of incubation (Fig. 3), suggesting that the cellular uptake and transfection of the shRNA is time-dependent. Additionally, since the AC–Cys(DBP)–Cys(mPEG)–AC micelles are able to expand in an acidic environment, these drug-loaded micelles can promptly escape from the acidic lysosomes, and subsequently be “cut” into pieces by the high level of GSH in the cytoplasm, achieving fast intracellular drug release. To further compare the internalization rate of the micelles, flow cytometry analysis was performed (Fig. 4), and DOX-micelles were used to replace the shRNA-micelles. From the flow cytometry results, we can see that the mean fluorescence intensity of the DOX group at 5 h is the highest and is approximately 7 times that of the group at 1 h, while there was approximately a 5-fold increase of fluorescence intensity found in the DOX/shRNA-micelles group and the DOX-micelles group, respectively. For the DOX group, the rapid increase of the fluorescence intensity may be because DOX can easily get into the cells through direct diffusion. Meanwhile, for DOX-loaded micelles, since the cellular internalization is a time-consuming process, the intensity of the fluorescence is relatively low and its increase is also relatively slow, compared to the DOX group.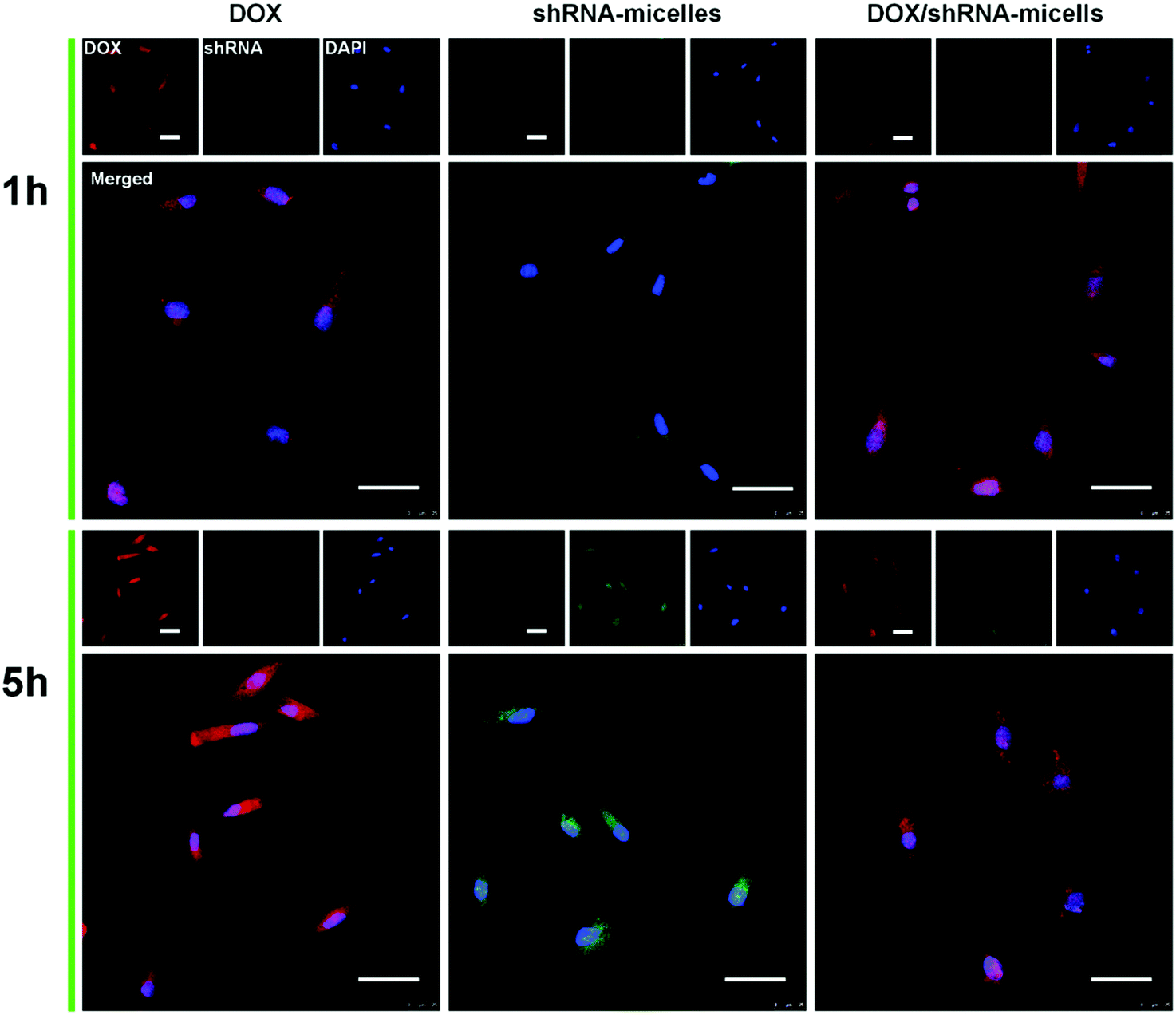 | ||
| Fig. 3 CLSM images of U87 cells incubated with DOX, shRNA-loaded micelles (shRNA-micelles) or DOX/shRNA-loaded micelles (DOX/shRNA-micelles) for 1 h or 5 h. Scale bar = 50 μm. Red fluorescence shows the DOX. Green fluorescence is expressed by cells after transfection of shRNA. Blue shows the cellular nucleus stained with DAPI. | ||
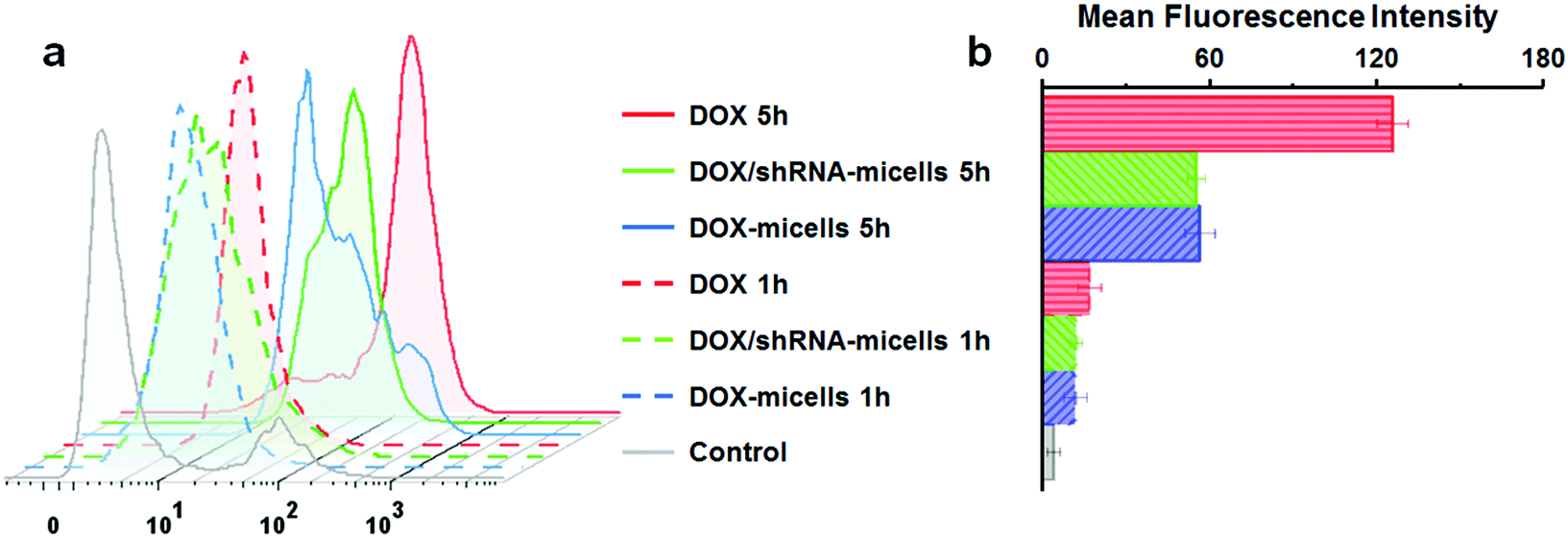 | ||
| Fig. 4 (a) Flow cytometry analysis of U87 cells incubated with different DOX formulations (DOX, DOX/PLK1 shRNA-micelles and DOX-micelles) for 1 h or 5 h (DOX equivalent dosage: 5 μg mL−1). (b) The corresponding intracellular mean fluorescence intensity of U87 cells incubated with different DOX formulations. | ||
For the shRNA transfection efficiency in U87 cells, flow cytometric analysis, shown in Fig. S6 (ESI†), offered quantitative data as follows: at 1 h, the transfection efficiencies of shRNA-PEI, shRNA-micelles and DOX/shRNA-micelles were 5.66%, 4.46% and 4.30%, respectively, while after 5 h of incubation, the transfection efficiencies were changed to 32.9% (shRNA-PEI), 24.3% (shRNA-micelles) and 27.5% (DOX/shRNA-micelles). These results suggest that though the transfection efficiency of PEI is the highest one, the prepared micelles have a comparable transfection efficiency. In addition, considering the cytotoxicity of PEI, our micelles would be an alternative carrier in gene delivery.
3.6 In vitro antitumor effect
To evaluate the in vitro anticancer effect of different drug formulations, U87 cells were further cultured with free DOX, DOX-micelles, DOX/shRNA-micelles, shRNA-micelles and shRNA-PEI at different DOX or shRNA concentrations. A dose-dependent effect on the U87 cell viability was present in all groups (Fig. 2b and c). For the DOX formulation, the IC50 values for DOX, DOX-micelles and DOX/shRNA-micelles are ∼1.7, ∼8.0 and ∼4.0 μg mL−1, respectively. For the shRNA formulation, the IC50 values for shRNA-PEI and shRNA-micelles are ∼0.38 and over 1.0 μg mL−1, respectively. The DOX possesses the lowest IC50 value, which may be due to the direct diffusion of DOX. The fact that the IC50 of the DOX/shRNA-micelles is half that of the DOX-micelles suggests that the DOX and shRNA have a synergetic effect on inhibiting the growth of cancer cells because the shRNA can down-regulate the PLK1 protein resulting in the suppression of mitosis in cancer cells. Notably, although the IC50 of shRNA-PEI is much lower than that of the shRNA-micelles, the inhibition effect of the cancer cell growth may be due to the cytotoxicity of the carrier polymer, PEI, which has been reported previously.45,46A cell apoptosis assay was applied to further verify the inhibition effect of the shRNA-PEI, shRNA-micelles, DOX-micelles and DOX/shRNA-micelles, as shown in Fig. 5. After a 24 h incubation period, the total apoptotic U87 cell populations induced by shRNA and shRNA-micelles were 44.6% and 20.5%, respectively, while those for the DOX-micelles and DOX/shRNA-micelles were 18.9% and 44.5%, respectively. It can be seen that the apoptosis rate of DOX-loaded micelles increased after introducing shRNA to the micelles, which is consistent with the IC50. In addition, though the shRNA-PEI has a similar inhibition effect as the DOX/shRNA-micelles, the polymeric micelles would be a better choice for a shRNA carrier, considering that PEI has relatively severe cytotoxicity compared to the micelles.
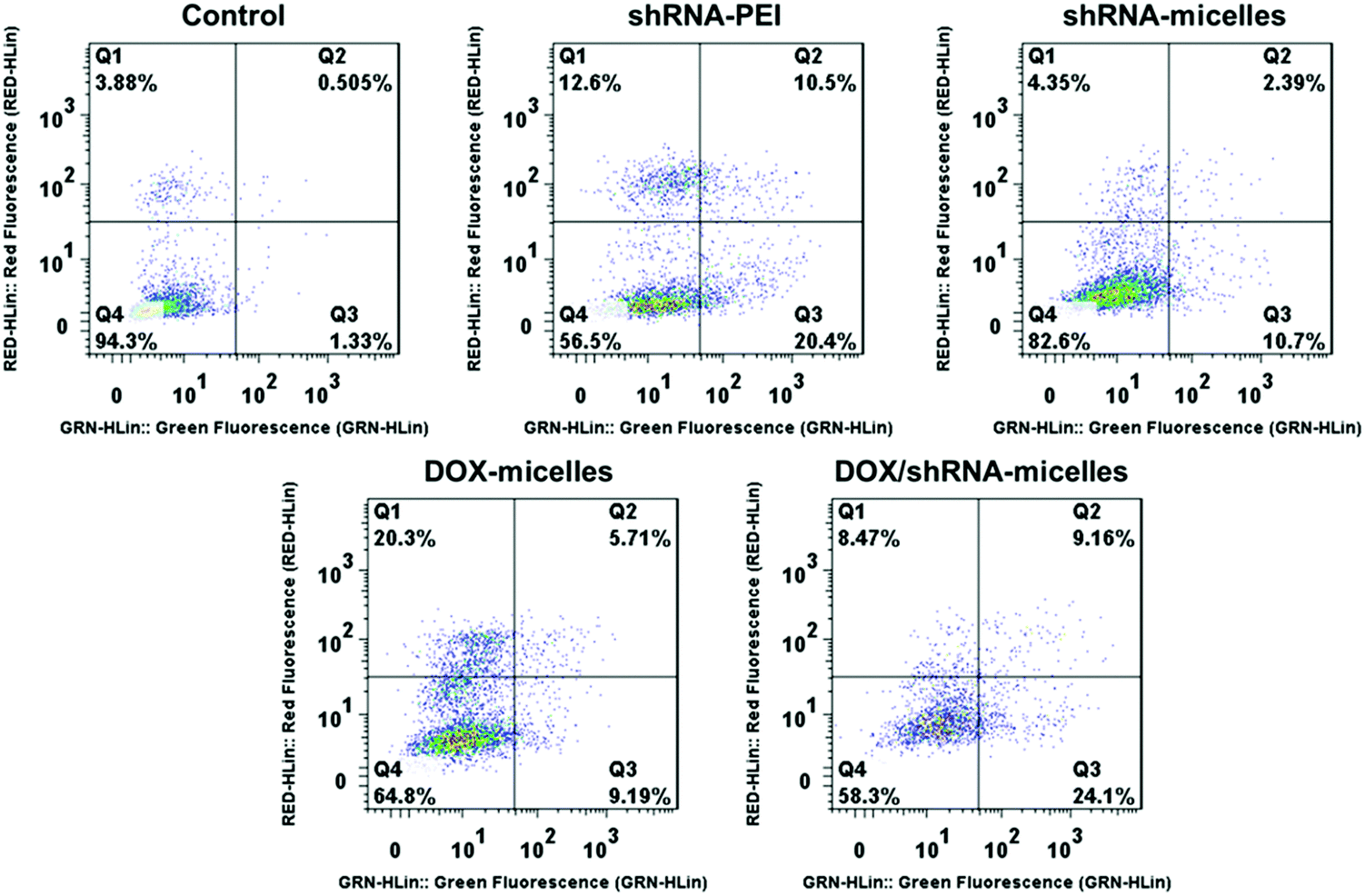 | ||
| Fig. 5 Apoptosis analysis of U87 cells induced by shRNA-PEI, shRNA-micelles, DOX-micelles and DOX/PLK1 shRNA-micelles after a 24 h incubation and stained with annexin V-FITC and PI for flow cytometry (FCM). DOX dosage: 5 μg mL−1; shRNA dosage: 0.4 μg mL−1. | ||
3.7 In vivo biodistribution
To directly visualize the distribution of the micelles in vivo, DOX and DOX-micelles were injected into U87 tumor-bearing nude mice, and the in vivo fluorescence intensity of DOX at different time intervals was monitored. Compared to the other groups, DOX-micelles accumulated more notably at the tumor site at all of the preset times as shown in Fig. 6a. Ex vivo imaging (Fig. 6b) also shows that the DOX fluorescence intensity in the tumor tissue of the DOX-micelles group is higher than that of other groups. Furthermore, the quantitative analysis (Fig. 6c) of the fluorescence intensities of the ex vivo tissues showed that the fluorescence intensity in the tumor tissue of the DOX-micelles group is about two times that of the DOX group. These results suggest that the micellar drug delivery system had a much stronger specificity to the target tumor in vivo. This can be attributed to two aspects: one is the ability of the micellar carrier to act as a shield using its bulky hydrophilic outer shell to avoid specific recognition by the reticuloendothelial system (RES),47 and the other is the enhanced penetration and retention (EPR) effect in tumor tissues, which have tortuous and leaky vasculatures.48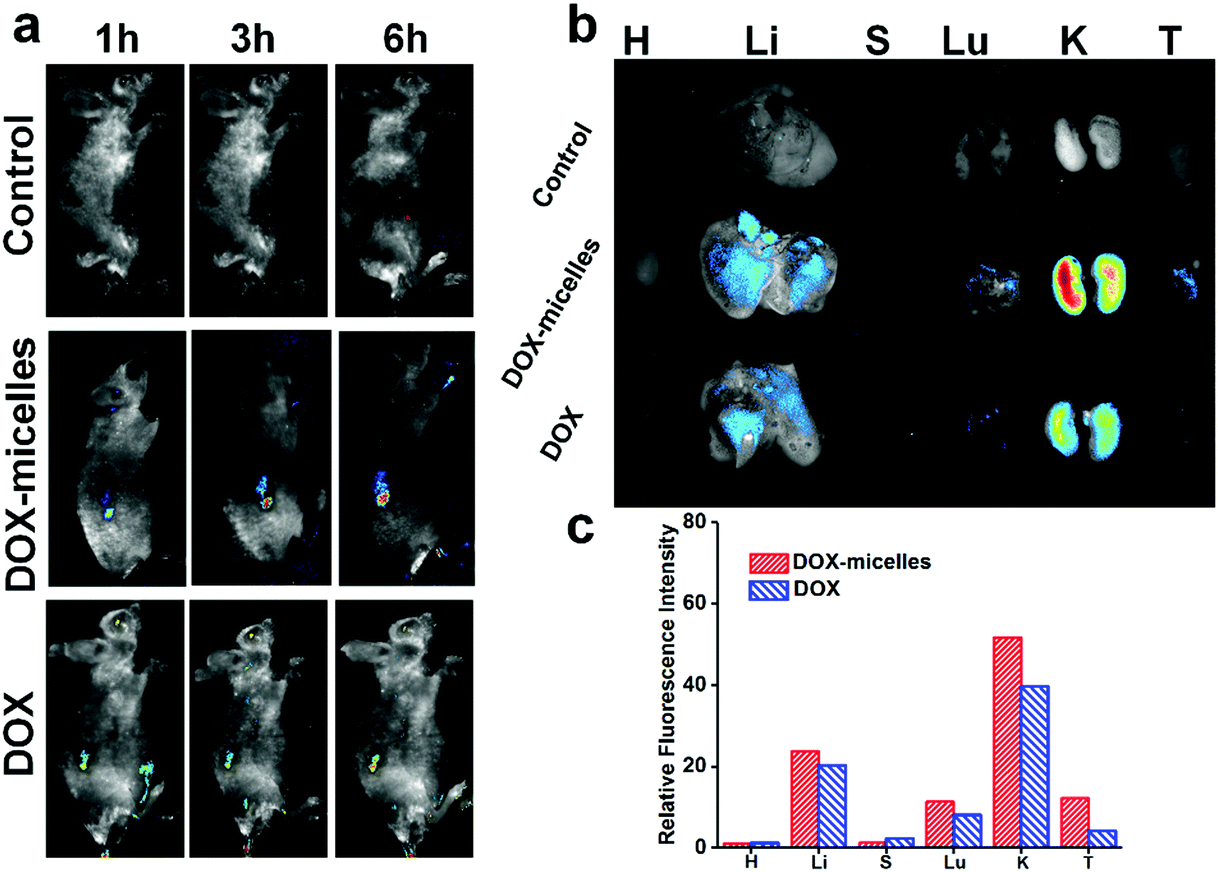 | ||
| Fig. 6 (a) In vivo DOX fluorescence images showing the DOX distribution in nude mice bearing subcutaneous U87 tumors after injection (DOX, DOX-micelles, and saline [control]) (DOX Dosage: 5 mg Dox per kg body weight) for 1 h, 3 h and 6 h. (b) Ex vivo fluorescence images showing the DOX distribution in organs and tumors extracted from the sacrificed mice after injection at 6 h. (c) The quantitative analysis of the fluorescence intensities of ex vivo tissues obtained by the Image-Pro Plus 6.0 software. | ||
3.8 In vivo antitumor effect
To evaluate the in vivo antitumor effect and toxicity of the various formulations, U87 tumor-bearing nude mice received intravenous administration of blank micelles, free DOX, DOX-micelles, shRNA-micelles and DOX/shRNA-micelles at 5 mg kg−1 equivalent DOX or 0.5 mg kg−1 equivalent shRNA, respectively. In Fig. 7a, it can be seen that the tumors in the nude mice from the different groups became larger with time. The tumor volume changes over 21 days are shown in Fig. 7b, and it can be seen that in all the micellar groups, shRNA-micelles (∼61 mm3), DOX-micelles (∼48 mm3) and DOX/shRNA-micelles (∼35 mm3) are smaller than the other groups including blank micelles (∼160 mm3) and DOX (∼128 mm3), and the DOX/shRNA-micelles group is the smallest. These changes in tumor volume demonstrate that compared to the blank micelles and the DOX group, the DOX-micelles, all of the shRNA-micelles and the DOX/shRNA-micelles possess the ability to suppress the growth of tumors. Specifically, the DOX/shRNA-micelle group showed approximately 1.3 times the tumor inhibition of the shRNA-micelle group and 1.1 times that of the DOX-micelle group, which means the DOX/shRNA-micelles displayed the most effective tumor growth inhibition, suggesting the synergetic anticancer effect of shRNA and DOX. Measurements of the body weight (Fig. 7c) and the survival rate (Fig. 7d) showed that the micellar groups were safer for the mice than free DOX, since the hydrophobic core of the micelles remarkably reduced the exposure of DOX to the normal tissues and thus decreased the toxicity of the chemotherapeutics. Furthermore, the tumor tissue histological study (Fig. 7e) is also supportive of the excellent therapeutic effect of the acidic and reduction-sensitive micelles containing DOX and shRNA simultaneously. After the treatment with the micellar formulations, we found that the tumor cells tend to be apoptotic and the amount of tumor cells gradually decreases, especially with the treatment of the DOX/shRNA-micelles. Thus, the DOX/shRNA-micelles were regarded as the most effective formulation in vivo. Additionally, the tumor microenvironment-sensitivity of the micelles helped the drug-loaded micelles to successfully escape the endo/lysosomes, which resulted in the delivery of the DOX to the nucleus and finally induced the apoptosis of the tumor cells. Additionally, the synergetic anticancer effect of the shRNA and DOX can effectively inhibit the growth of tumor cells. These results indicate that micelles from AC–Cys(DBP)–Cys(mPEG)–AC can be a suitable nanocarrier in anticancer drug delivery and the DOX/shRNA-micelles have excellent superiority in cancer therapy.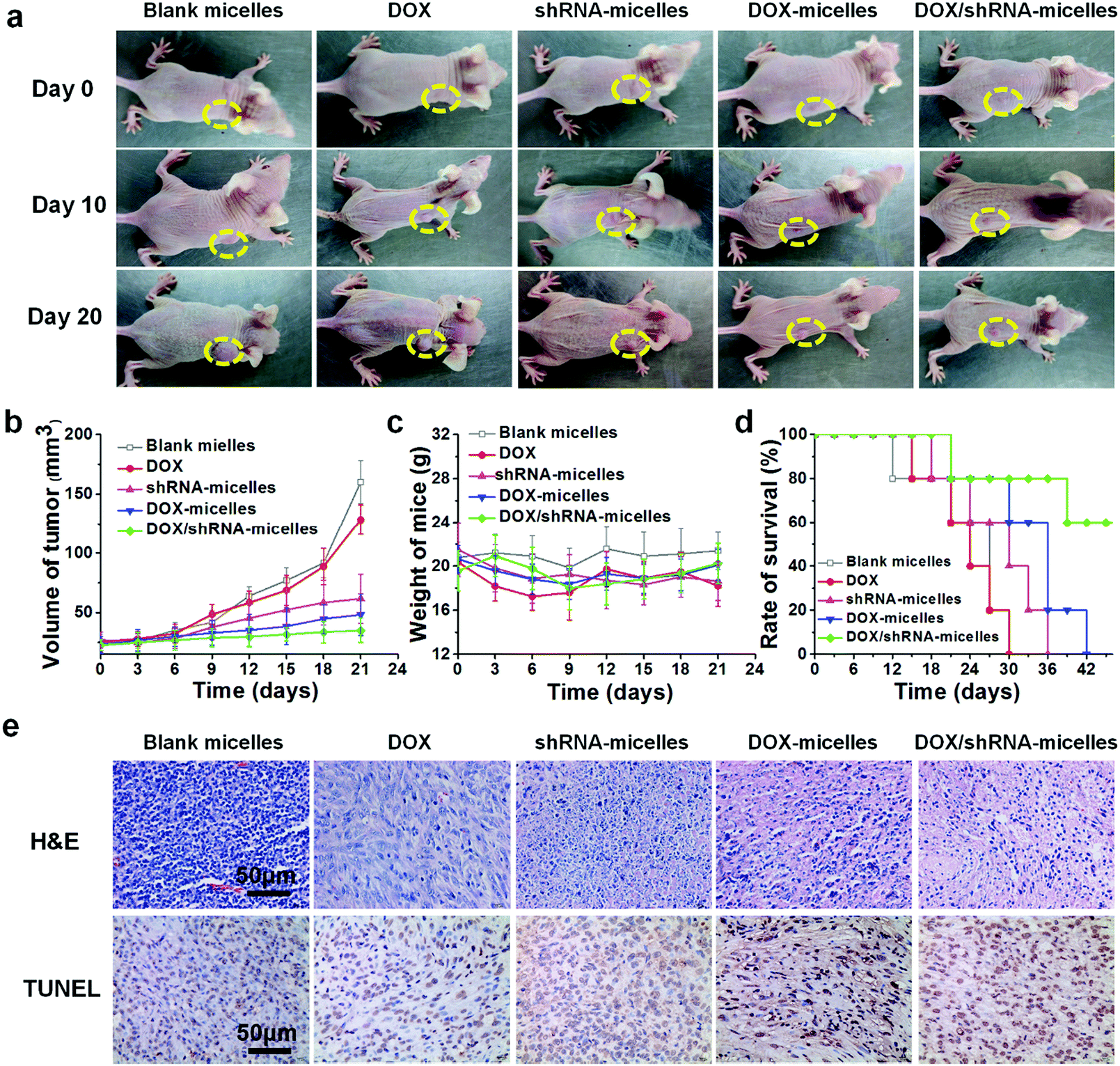 | ||
| Fig. 7 In vivo therapeutic effect and systemic toxicity of blank micelles, free DOX, shRNA-micelles, DOX-micelles and DOX/PLK1 shRNA-micelles injected into nude mice bearing subcutaneous U87 tumors at a DOX-dose of 5 mg kg−1 and shRNA-dose of 0.4 μg kg−1. (a) Photos of the representative tumor-bearing nude mice in different groups at day 0, day 10 and day 20 during the treatment. (b) Variations of tumor volume, (c) the body weight and (d) survival rate of tumor-bearing nude mice during 21 days after the first injection of different drug formulations. (e) Histological analyses of the tumor sections (14 days after the first injection). | ||
Western blotting analysis and RT-PCR were performed to determine the mechanism of the tumor suppression in vivo. The polo-like kinase 1, PLK1, was analyzed by RT-PCR. Compared to the other groups, an enhancement of the PLK1 expression can be found in the DOX/shRNA group. In addition, the PLK1, proapoptotic gene caspase-3 and antiapoptotic gene BCL2 expressions were further evaluated by western blot analysis as shown in Fig. 8c. We can see that the treatment of the DOX/shRNA group led to the most significant up-regulation of PLK1 and the caspase-3 protein and down-regulation of the BLC2 protein. Together with the RT-PCR, these results indicate that the DOX/shRNA-loaded tumor microenvironment-sensitive polymer micelles induced apoptosis through an enhanced DNA damage-mediated pathway.49
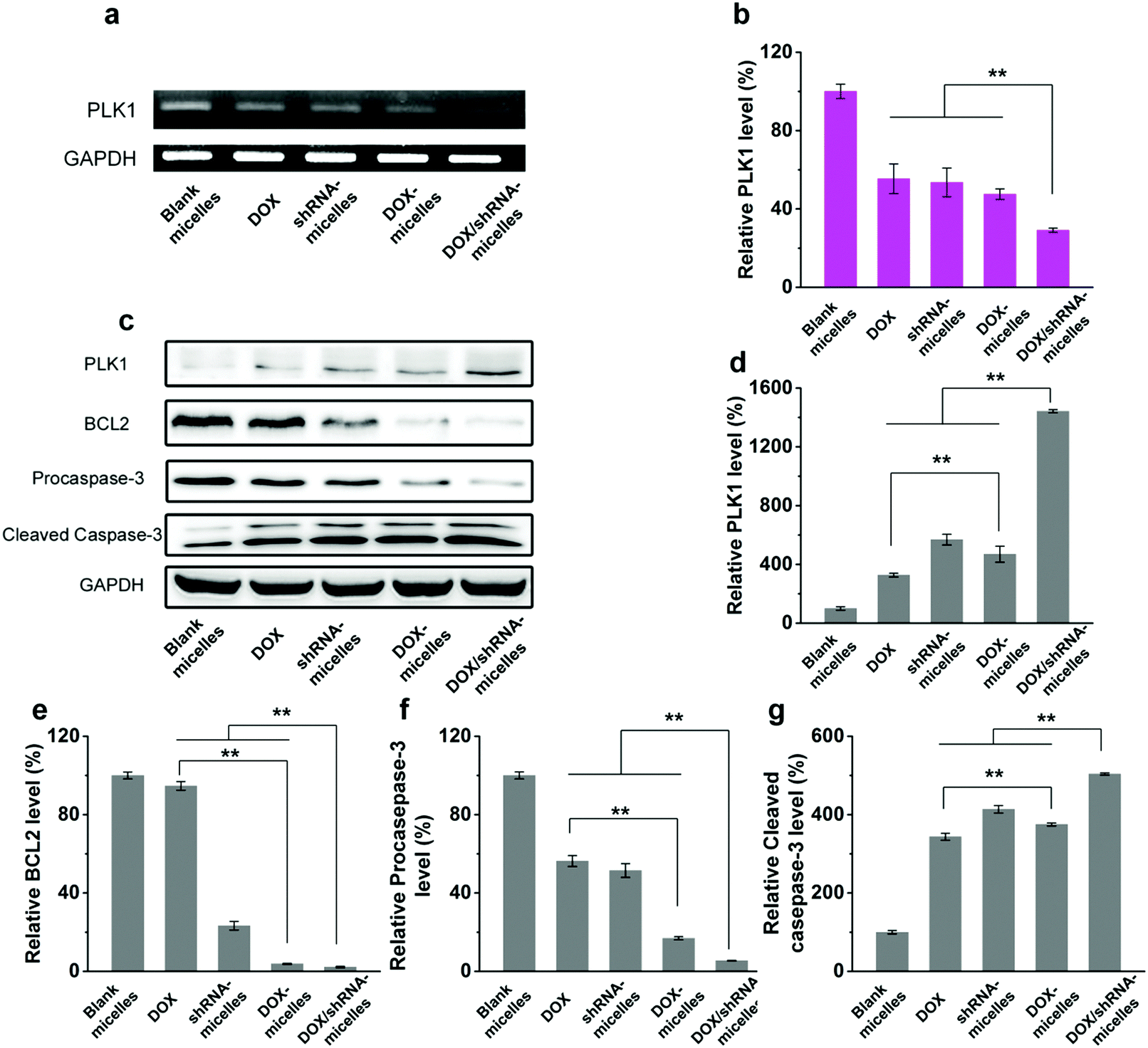 | ||
| Fig. 8 Inhibition of endogenous PLK1 mRNA and PLK1 protein expression with tissue of U87 tumor-bearing nude mice in vivo. (a) Representative PLK1 mRNA was analyzed by semiquantitative RT-PCR. GADPH was used an internal control. (b) PLK1 mRNA levels were quantitative by RT-PCR. (c) Representative PLK1, BCL2, procaspase-3, and cleaved caspase-3 protein was analyzed by western blot. GADPH was used as an internal control. (d–g) PLK1, BCL2, procaspase-3, and cleaved caspase-3 protein relative to GADPH were determined by ImageJ. The results are expressed as the means (±SD) from independent mean gray value analysis. Asterisks (*) denote statistically significant differences (**p < 0.001) calculated by a one-way ANOVA test. | ||
4 Conclusions
In summary, an acidic- and redox-sensitive copolymer was synthesized successfully by using a “one-pot method”. The micelles formed by self-assembly from the copolymer possess a tumor microenvironment-sensitivity and can be triggered to rapidly release the encapsulated drugs. In vitro antitumor analysis reveals that the DOX/shRNA micelles are effective in inhibiting the growth of tumor cells due to the synergetic therapeutic effect of DOX and shRNA released rapidly in a tumor microenvironment. The mechanism of the synergism is that the released DOX molecules integrate with nuclear DNA, causing DNA damage, and the PLK1-specific shRNA down-regulates the expression of PLK1. In vivo antitumor activity also demonstrates that these micelles are safer and more therapeutic than the free DOX and single drug-loaded micelles. Therefore, the micelles loading dual therapeutic agents have great potential as a nanocarrier for cancer therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the National Natural Science Foundation of China (Grant No. 51373138, 21574105, and 51725303), the National Key Research and Development Program of China (2017YFB0702600, and 2017YFB0702602), the Sichuan Province Youth Science and Technology Innovation Team (Grant No. 2016TD0026) and the Fundamental Research Funds for the Central Universities (2682017ZT02).References
- B. Newland, Y. Zheng, Y. Jin, M. Abu-Rub, H. Cao, W. Wang and A. Pandit, J. Am. Chem. Soc., 2012, 134, 4782–4789 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Delivery Rev., 2013, 65, 71–79 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- C. Deng, Y. Jiang, R. Cheng, F. Meng and Z. Zhong, Nano Today, 2012, 7, 467–480 CrossRef CAS.
- C. Wang, Z. Wang and X. Zhang, Acc. Chem. Res., 2012, 45, 608–618 CrossRef CAS PubMed.
- T. M. Allen, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- M. E. Fox, F. C. Szoka and J. M. J. Fréchet, Acc. Chem. Res., 2009, 42, 1141–1151 CrossRef CAS PubMed.
- A. Albanese, P. S. Tang and W. C. W. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed.
- Q. Qu, Y. Wang, L. Zhang, X. Zhang and S. Zhou, Small, 2016, 12, 1378–1390 CrossRef CAS PubMed.
- X. Guo, C. Shi, G. Yang, J. Wang, Z. Cai and S. Zhou, Chem. Mater., 2014, 26, 4405–4418 CrossRef CAS.
- Y. Wang, L. Zhang, X. Zhang, X. Wei, Z. Tang and S. Zhou, ACS Appl. Mater. Interfaces, 2016, 8, 5833–5846 CAS.
- X. Guo, X. Wei, Y. Jing and S. Zhou, Adv. Mater., 2015, 27, 6450–6456 CrossRef CAS PubMed.
- W. Gao, J. M. Chan and O. C. Farokhzad, Mol. Pharmaceutics, 2010, 7, 1913–1920 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- J. A. Hubbell, Science, 2003, 300, 595–596 CrossRef CAS PubMed.
- R. P. Johnson, Y. Jeong, E. Choi, C. Chung, D. H. Kang, S. Oh, H. Suh and I. Kim, Adv. Funct. Mater., 2012, 22, 1058–1068 CrossRef CAS.
- X. Liu, Y. Chen, H. Li, N. Huang, Q. Jin, K. Ren and J. Ji, ACS Nano, 2013, 7, 6244–6257 CrossRef CAS PubMed.
- X. Guo, C. Shi, J. Wang, S. Di and S. Zhou, Biomaterials, 2013, 34, 4544–4554 CrossRef CAS PubMed.
- C. Shi, X. Guo, Q. Qu, Z. Tang, Y. Wang and S. Zhou, Biomaterials, 2014, 35, 8711–8722 CrossRef CAS PubMed.
- J. Zhang, Z. Yuan, Y. Wang, W. Chen, G. Luo, S. Cheng, R. Zhuo and X. Zhang, J. Am. Chem. Soc., 2013, 135, 5068–5073 CrossRef CAS PubMed.
- J. Wang, G. Yang, X. Guo, Z. Tang, Z. Zhong and S. Zhou, Biomaterials, 2014, 35, 3080–3090 CrossRef CAS PubMed.
- H. Sun, B. Guo, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomaterials, 2009, 30, 6358–6366 CrossRef CAS PubMed.
- K. Wang, G. Luo, Y. Liu, C. Li, S. Cheng, R. Zhuo and X. Zhang, Polym. Chem., 2012, 3, 1084 RSC.
- L. Tang, Y. Wang, Y. Li, J. Du and J. Wang, Bioconjugate Chem., 2009, 20, 1095–1099 CrossRef CAS PubMed.
- X. Wei, Y. Wang, X. Xiong, X. Guo, L. Zhang, X. Zhang and S. Zhou, Adv. Funct. Mater., 2016, 26, 8266–8280 CrossRef CAS.
- Z. Qiao, D. Zhang, C. Hou, S. Zhao, Y. Liu, Y. Gao, N. Tan and H. Wang, J. Mater. Chem. B, 2015, 3, 4514–4523 RSC.
- L. Wei, C. Cai, J. Lin and T. Chen, Biomaterials, 2009, 30, 2606–2613 CrossRef CAS PubMed.
- J. Lin, J. Zhu, T. Chen, S. Lin, C. Cai, L. Zhang, Y. Zhuang and X. S. Wang, Biomaterials, 2009, 30, 108–117 CrossRef CAS PubMed.
- S. Aryal, C. M. Hu and L. Zhang, Small, 2010, 6, 1442–1448 CrossRef CAS PubMed.
- H. Meng, M. Liong, T. Xia, Z. Li, Z. Ji, J. I. Zink and A. E. Nel, ACS Nano, 2010, 4, 4539–4550 CrossRef CAS PubMed.
- L. Chen, T. Jiang, C. Cai, L. Wang, J. Lin and X. Cao, Adv. Healthcare Mater., 2014, 3, 1508–1517 CrossRef CAS PubMed.
- H. Osada and S. Simizu, Nat. Cell Biol., 2000, 2, 852–854 CrossRef PubMed.
- M. R. Smith, M. L. Wilson, R. Hamanaka, D. Chase, H. Kung, D. L. Longo and D. K. Ferris, Biochem. Biophys. Res. Commun., 1997, 234, 397–405 CrossRef CAS PubMed.
- Z. R. Cohen, S. Ramishetti, N. Peshes-Yaloz, M. Goldsmith, A. Wohl, Z. Zibly and D. Peer, ACS Nano, 2015, 9, 1581–1591 CrossRef CAS PubMed.
- S. Sharma, K. C. Basavaraju, A. K. Singh and D. Kim, Org. Lett., 2014, 16, 3974–3977 CrossRef CAS PubMed.
- L. Wang, X. Xu, C. Hong, D. Wu, Z. Yu and Y. You, Chem. Commun., 2014, 50, 9676 RSC.
- Y. Wang, Y. Lin, Z. Qiao, H. An, S. Qiao, L. Wang, R. P. Y. J. Rajapaksha and H. Wang, Adv. Mater., 2015, 27, 2627–2634 CrossRef CAS PubMed.
- R. Mo, Q. Sun, J. Xue, N. Li, W. Li, C. Zhang and Q. Ping, Adv. Mater., 2012, 24, 3659–3665 CrossRef CAS PubMed.
- F. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180–2198 CrossRef CAS PubMed.
- J. Liu, Y. Pang, W. Huang, X. Huang, L. Meng, X. Zhu, Y. Zhou and D. Yan, Biomacromolecules, 2011, 12, 1567–1577 CrossRef CAS PubMed.
- W. Yuan, T. Shen, J. Wang and H. Zou, Polym. Chem., 2014, 5, 3968 RSC.
- K. Wang, Y. Liu, W. Yi, C. Li, Y. Li, R. Zhuo and X. Zhang, Soft Matter, 2013, 9, 692–699 RSC.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef CAS PubMed.
- C. Brunot, L. Ponsonnet, C. Lagneau, P. Farge, C. Picart and B. Grosgogeat, Biomaterials, 2007, 28, 632–640 CrossRef CAS PubMed.
- D. Putnam, C. A. Gentry, D. W. Pack and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1200–1205 CrossRef CAS.
- J. Geng, K. Li, D. Ding, X. Zhang, W. Qin, J. Liu, B. Z. Tang and B. Liu, Small, 2012, 8, 3655–3663 CrossRef CAS PubMed.
- D. J. Bharali, D. W. Lucey, H. Jayakumar, H. E. Pudavar and P. N. Prasad, J. Am. Chem. Soc., 2005, 127, 11364–11371 CrossRef CAS PubMed.
- E. Shaulian and M. Karin, Oncogene, 2001, 20, 2390–2400 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tb02160g |
| ‡ These authors contributed equally to this work |
| This journal is © The Royal Society of Chemistry 2018 |