
Construction of giant glycosidase inhibitors from iminosugar-substituted fullerene macromonomers†
M. Isabel
García-Moreno,  c
José M.
García Fernández,
d
Philippe
Compain,
*b
Carmen
Ortiz Mellet
*c
and
Jean-François
Nierengarten
*a
c
José M.
García Fernández,
d
Philippe
Compain,
*b
Carmen
Ortiz Mellet
*c
and
Jean-François
Nierengarten
*a
c
José M.
García Fernández,
d
Philippe
Compain,
*b
Carmen
Ortiz Mellet
*c
and
Jean-François
Nierengarten
*a
Abstract
An ultra-fast synthetic procedure based on grafting of twelve fullerene macromonomers onto a fullerene hexa-adduct core was used for the preparation of a giant molecule with 120 peripheral iminosugar residues. The inhibition profile of this giant iminosugar ball was evaluated against various glycosidases. In the particular case of the Jack bean α-mannosidase, a dramatic enhancement of the glycosidase inhibitory effect was observed for the giant molecule with 120 peripheral subunits as compared to that of the corresponding mono- and dodecavalent model compounds.
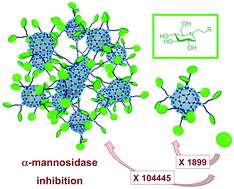
- This article is part of the themed collections: 2017 Journal of Materials Chemistry B HOT Papers and Carbon Nanostructures in Biology and Medicine
 Please wait while we load your content...
Please wait while we load your content...

