
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold–iron oxide dimers for magnetic hyperthermia: the key role of chloride ions in the synthesis to boost the heating efficiency†
P.
Guardia‡
ab,
S.
Nitti‡
a,
M. E.
Materia
a,
G.
Pugliese
a,
N.
Yaacoub
c,
J.-M.
Greneche
c,
C.
Lefevre
d,
L.
Manna
a and
T.
Pellegrino
*a
aIstituto Italiano di Tecnologia, Via Morego 30, 16163 Genova, Italy. E-mail: teresa.pelegrino@iit.it
bCentro de Tecnologia Quimica de Cataluña, Carrer de Marcel·lí Domingo s/n, 43007 Tarragona, Spain
cInstitut des Molécules et Matériaux du Mans IMMM UMR CNRS 6283, Université du Maine, Avenue Olivier Messiaen, F-72085 Le Mans Cedex, France
dInstitut de Physique et de Chimie des Matériaux de Strasbourg 23, rue du Loess, BP 43 67034 Strasbourg Cedex 2, France
First published on 28th April 2017
Abstract
With the aim of producing Au–FexOy dimers with outstanding heating performances under magnetic hyperthermia conditions applicable to human patients, here we report two synthesis routes, a two-pot and a one-pot method. The addition of chloride ions and the absence of 1,2-hexadecanediol (HDDOL), a commonly used chemical in this synthesis, are the key factors that enable us to produce dimers at low temperature with crystalline iron oxide domains in the size range between 18–39 nm that is ideal for magnetic hyperthermia. In the case of two-pot synthesis, in which no chloride ions are initially present in the reaction pot, dimers are obtained only at 300 °C. In order to lower the reaction temperature to 200 °C and to tune the size of the iron oxide domain, the addition of chloride ions becomes the crucial parameter. In the one-pot method, the presence of chloride ions from the start of the synthesis (as counter ions of the gold salt precursor) enables a prompt formation of dimers directly at 200 °C. In this case, the reaction time is the main parameter used to tune the iron oxide size. A record value of specific absorption rates (SARs) up to 1300 W gFe−1 at 330 kHz and 24 kA m−1 was measured for dimers with an iron oxide domain of 24 nm in size.
Introduction
The application of magnetic hyperthermia (MH) in clinics to treat tumor requires highly efficient magnetic nanoparticles (NPs), capable of rising the tumor temperature with spatial and temporal control at the lowest possible NP dose, in order to reduce systemic toxicity.1–4 Along this direction, iron oxide nanocubes with cube-edge sizes between 18 and 35 nm have shown outstanding specific absorption rate (SAR) values.5,6 Also, hard-soft core–shell magnetic NPs have remarkably high SAR values, but the toxicity of some of their ions (i.e. cobalt and nickel ferrites) requires accurate studies prior to their use in clinics.7,8 Instead, much less is known about the SAR performance of heterostructures that merge an iron oxide NP with other materials in one single nanostructure. The most relevant example is given by Au–iron oxide dimers made of Au NPs attached to an iron oxide domain by a small interface. After the pioneering works of Sun et al. on the synthesis of Au–Fe2O3 heterodimers and their metal-alloyed counterparts,9–11 along with a more recent study on the mechanism of dimer growth,12 different uses, from catalysis13–15 to biomedicine, were pursued.16The gold domain can be exploited for two-photon imaging, surface enhanced Raman scattering and computer tomography,10,17 while the iron oxide NPs can enable molecular resonance imaging,16 or they can serve as heat mediators for MH. Only one work to the best of our knowledge has thus far been reported on the hyperthermia performance of dimers (made of 2 nm Au and 14 nm iron oxide NPs), however with poor SAR values.9 Whereas in catalysis tuning the size of the Au domain can raise the catalytic activity of the dimers, for MH size tuning of the iron oxide NPs in the 20–30 nm range is the key to boost the heating performance.18 So far, dimers with large iron oxide NPs (>20 nm) were prepared either with a poor control over the size or simply they were not designed and tested for MH (high SAR, high crystallinity, etc.).9,10,12,16 Here, we report two protocols for the synthesis of gold–iron oxide NP dimers with a tunable iron oxide domain size, in both cases using standard air-free techniques.
In a “two-pot” approach, we used pre-synthesized Au NPs as seeds on which iron oxide was nucleated; instead in the “one-pot” method Au NPs were first nucleated in situ followed by the growth of the iron oxide domain (Fig. 1A and ESI†). We found that the presence of chloride ions (Cl−) in the reaction flask reduces the reaction temperatures at which iron oxide nucleates on top of the Au NPs, enabling a tunable growth of the iron oxide domain with sizes exceeding 20 nm (up to 39 nm in this work). Moreover, the type of Cl chemical and the Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cl ratio are the parameters that were exploited to tune the size and shape of the iron oxide domain. Finally, the absence of 1,2-hexadecanediol (HDDOL), a commonly used surfactant in the synthesis, favours the growth of dimers with larger iron oxide domains as required for MH.
:Cl ratio are the parameters that were exploited to tune the size and shape of the iron oxide domain. Finally, the absence of 1,2-hexadecanediol (HDDOL), a commonly used surfactant in the synthesis, favours the growth of dimers with larger iron oxide domains as required for MH.
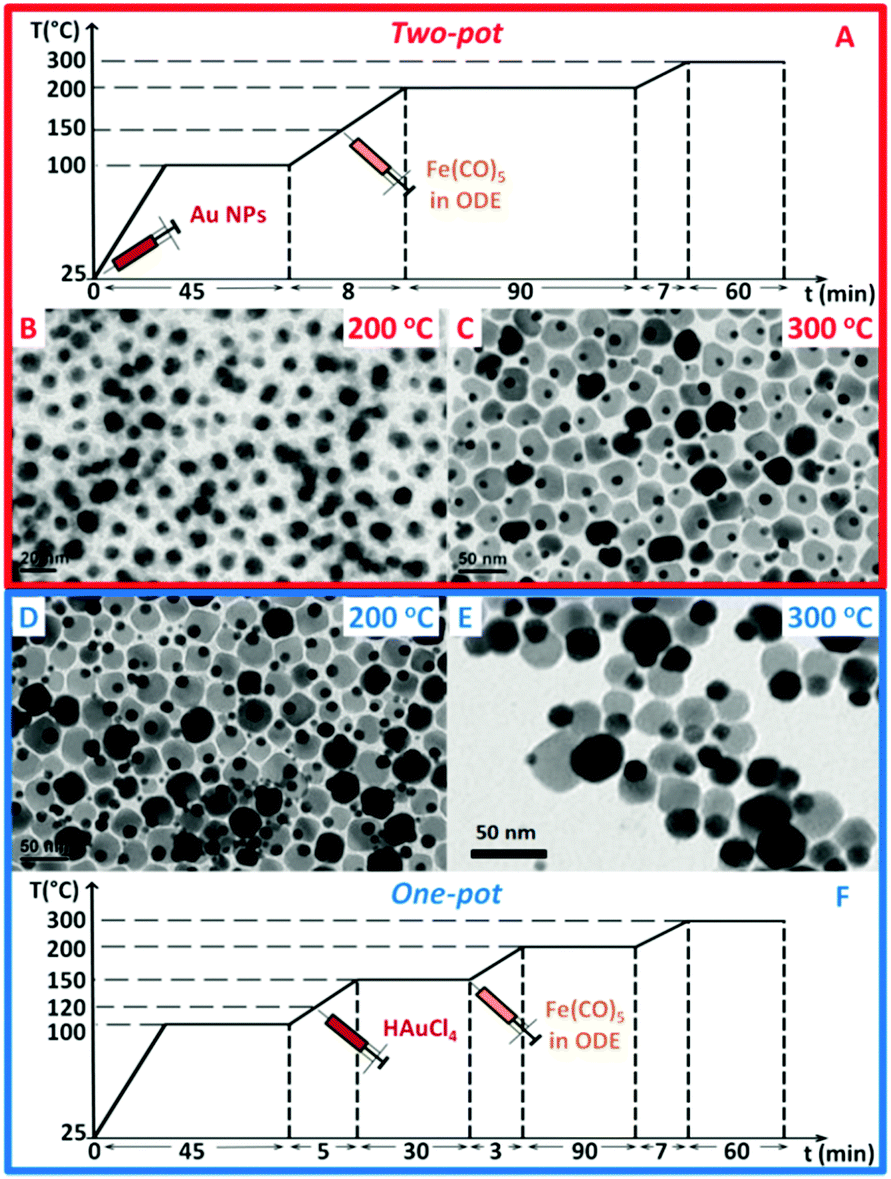 | ||
| Fig. 1 Temperature profiles for the synthesis by one-pot (A) or two-pot (F) methods of gold–iron oxide dimers. In the two-pot approach, starting with pre-formed Au NPs present in the reaction flask since the beginning of the reaction (Fig. S1, ESI†), upon iron precursor injection, dark spots on the Au NPs appear after 90 min at 200 °C (B); and only at 300 °C dimers are formed (C). In the one-pot approach, the gold precursor (HAuCl4) is instead injected at 120 °C and Au NPs nucleate in situ. In this case dimers are already formed at 200 °C (D) and they grow further at 300 °C (E). | ||
Materials and methods
Chemicals
1-Octadecene (90%), oleylamine (70%), oleic acid (90%), iron pentacarbonyl Fe(CO)5 (>99.99%), tetrachloroauric(III) acid HAuCl4·3H2O (≥99.9%), 1,2-hexadecanediol (90%) (1,2-HDDOL), 1,2-dodecanediol (90%), 1,2-dichloroethane (≥99.0%), hydrogen chloride solution (2.0 M in diethyl ether), hydrochloric acid (HCl, ≥37%), tetrabutylammonium chloride (≥97.0%), 1-chlorooctadecane (96%), chloroform (ACS grade), 2-propanol (ACS grade), and ethanol (absolute, ACS grade) were purchased from Sigma Aldrich and used as received.Synthesis of 9 nm Au NPs
In a 100 mL three-neck flask; 20 mL of 1-octadecene, 3 mL of oleylamine and 1 mL of oleic acid were mixed and degassed at 120 °C for 30 minutes. Meanwhile, a fresh solution of the gold precursor was prepared by mixing 0.12 mmol (40 mg) of HAuCl4·3H2O, 0.5 mL oleylamine and 5 mL 1-octadecene, and vortexed until the gold salt was completely dissolved. Under nitrogen flow, the gold solution was injected at 120 °C into the flask and heated up to 150 °C. After 30 minutes at 150 °C, the solution was cooled down to room temperature and 70 mL of 2-propanol and 10 mL of ethanol were added. The solution was then centrifuged at 5000 rpm for 20 minutes, the supernatant was discarded and the precipitate was redissolved in 4 mL of hexane. An additional washing step was carried out by the addition of 20 mL of 2-propanol and 5 mL of ethanol followed by centrifugation at 5000 rpm for 20 minutes. The final Au NPs were re-dissolved in 4 mL of hexane and centrifuged again at 1000 rpm for 5 minutes in order to remove possible aggregates. The final supernatant was kept and protected from light at 5 °C for further use.“Two-pot 200” gold–iron oxide dimer synthesis
In a 100 mL three-neck flask, 20 mL of 1-octadecene, 3 mL of oleylamine and 1 mL of oleic acid were degassed at 120 °C for 30 minutes. The solution was then cooled to 65 °C and a hexane solution of premade Au NPs (8.8 mg of Au mass, 0.045 mmol Au) was injected under nitrogen flow. The solution was degassed for 60 minutes at 65 °C and then heated up to 150 °C under nitrogen flow. At 150 °C, an iron precursor solution containing 1 mL of 1-octadecene and 80 μL of Fe(CO)5 (0.6 mmol) was injected. The reaction temperature was then increased to 200 °C at a heating rate of 7 °C min−1 and kept at this temperature for 90 minutes. Finally, the solution was cooled down to room temperature and dimers were washed and stored as described later.“Two-pot 300” gold–iron oxide dimer synthesis
As for the two-pot 200 synthesis, the same protocol was followed with the only difference that, after annealing at 200 °C for 90 minutes, the temperature was further increased to 300 °C (7 °C min−1) and kept at this temperature for 60 minutes. After cooling down the flask to room temperature dimers were collected and washed as described later.Chloride-mediated synthesis of gold–iron oxide dimers using a “two-pot 200” approach
As for the two-pot 200 synthesis; 20 mL of 1-octadecene, 3 mL of oleylamine and 1 mL of oleic acid were mixed and degassed at 120 °C for 30 minutes. The solution was then cooled down to 65 °C and a solution containing premade Au NPs (8.8 mg of Au in 5 mL of hexane) was injected under nitrogen flow. Keeping the temperature constant (65 °C) the solution was set under vacuum for 60 minutes to completely remove hexane. Then, a given amount of a 2 M solution of HCl in diethyl ether (for instance 120 μL, 0.24 mmol of HCl) was injected under nitrogen flow prior to heating the solution up to 150 °C. Once the temperature was reached, a solution containing 80 μL of Fe(CO)5 (0.6 mmol) in 1 mL of 1-octadecene was injected and the temperature was increased up to 200 °C at a heating rate of 7 °C min−1. After 90 minutes at this temperature, the solution was cooled down to room temperature and washed as described later. Note that the Cl precursor can be switched to other compounds such as 1,2-dichloroethane.“One-pot 200” gold–iron oxide dimer synthesis
The one-pot method allows us to in situ produce Au NPs and then grow the iron oxide NPs. 20 mL of 1-octadecene, 3 mL of oleylamine and 1 mL of oleic acid were mixed in a 100 mL three-neck flask and degassed at 120 °C for 30 minutes. Then a freshly prepared gold solution (0.12 mmol (40 mg) of HAuCl4·3H2O in 0.5 mL oleylamine and 5 mL 1-octadecene) was injected at 120 °C under a nitrogen flow blanket. The reaction mixture was then heated up to 150 °C and kept at this temperature for 30 minutes. An aliquot was withdrawn in order to control the initial Au NPs (data not shown). Subsequently, a solution containing 1 mL of 1-octadecene and 80 μL of Fe(CO)5 (0.6 mmol) was injected at 150 °C and the temperature was increased to 200 °C at a heating rate of 7 °C min−1. After 90 minutes at 200 °C, the solution was cooled down to room temperature. In a series of experiments, the iron oxide size was controlled by stopping the reaction at different times (30, 60 or 90 minutes). To check the effect of the 1,2-HDDOL surfactant, the same one-pot 200 synthesis was repeated following the same protocol with the only difference that 1,2-HDDOL (2.58 g 10 mmol) was added as a surfactant to the initial reaction flask. The one-pot 200 procedure could be simplified following the same protocol as described above but injecting the gold salt at the beginning of the reaction. In this case Au NPs are already formed after the vacuum step.“One-pot 300” synthesis of gold–iron oxide dimers
Following exactly the same protocol as described for the one-pot 200, the solution was further heated up to 300 °C (7 °C min−1) and kept at this temperature for 60 minutes.Dimer sample washing procedure
For all the dimer syntheses reported here the washing procedure used was as follows: after keeping the reaction mixture at high temperature (300 or 200 °C), the mixture was cooled down to room temperature slowly, and 80 mL of 2-propanol were added to the crude (the magnetic stirrer was washed with care to recover the NPs sticked on the surface). After centrifugation at 5000 rpm for 20 minutes the supernatant was discarded and the precipitate was re-dissolved in 4 mL of chloroform. An additional washing step was performed by adding 20 mL of 2-propanol followed by centrifugation at 5000 rpm for 20 minutes. The final precipitate containing the dimers was re-dissolved in 4 mL of chloroform and protected from light at 5 °C.Transfer of gold–iron oxide dimers in polar solvents
To transfer dimers into water an already reported ligand exchange procedure was used with some slight modifications.19 In a 40 mL vial, 8 mL of dimer solution in chloroform with an iron concentration of 1 g L−1 were mixed with 12.2 mL of a chloroform solution of gallol polyethylene glycol (GA-PEG) (Mw = 3000 kDa, 0.05 M) corresponding to 1000 molecules per nm2, and 1.22 mL of triethylamine. The whole solution was stirred overnight at room temperature and evaporated under reduced pressure at 40 °C. The dried solution was dissolved in 10 mL of methanol, transferred in a separating funnel and 5 mL of hexane were added. After emulsification by shaking, the two phases were allowed to separate. The methanol phase containing the gold–iron oxide dimers bearing GA-PEG was collected and the hexane phase was discarded. The extraction was repeated two more times to remove the free PEG ligand in solution. The dimer solution in methanol was dried under reduced pressure at 40 °C; then the nanoparticles were re-dissolved in 10 mL of water and the excess of GA-PEG was removed by dialysis against de-ionized water (5 L) using a cellulose membrane tubing (molecular weight cut-off, MWCO of 50 kDa). The sample was dialysed overnight at room temperature. Finally, the solution was concentrated by centrifugation in a 2 mL centrifuge filter tube (MWCO 100 kDa).Etching procedure to remove the gold domain from dimers
The removal of the gold domain from the gold–iron oxide dimers was performed by strong oxidation of gold with Lugol's solution (a water iodine solution made of 2% (w/v) of I2 and 4% (w/v) of KI). To 600 μL of dimers in water (Au concentration of 0.2 μM) 6 mL of a 50 times diluted Lugol's solution were added. The solution was shaken at 60 °C for 1 h. Soon after the reaction mixture was diluted to 10 mL with milliQ water and sonicated for a few minutes. To remove the oxidizing agent and the free gold ions several washes with fresh water were performed by collecting the empty dimers on a permanent magnet (0.3 T).Dynamic light scattering (DLS)
Dynamic light scattering (DLS) measurements were performed on a Zetasizer Nano ZS90 (Malvern) equipped with a 4.0 mW He–Ne laser operating at 633 nm and an avalanche photodiode detector. Measurements were conducted using a ZEN0112-low volume disposable sizing cuvette in water. The refractive index was set at 1.330 for water and the viscosity value was set at 0.8872 cP. The measurements were performed with 173° backscatter (NIBS default) as the angle of detection, with an automatic scan time and three scans per measurement.Magnetic characterization
Magnetic characterization was carried out using a superconducting quantum interference device (SQUID) from Quantum Design. Magnetization curves were measured from −80 to +80 kOe at 5 K and 300 K upon zero field cooling (ZFC). Zero field cooled (ZFC) and field cooled (FC) curves were recorded to measure the thermal dependence of the magnetization. Samples were prepared by drop casting a solution of gold–iron oxide dimers onto a Teflon film and the amount of materials measured was evaluated by elemental analysis.Hyperthermia measurements
A commercially available set-up (DM100 Series, nanoScale Biomagnetics Corp.) was used in order to test the hyperthermia performance of gold–iron oxide dimers. 500 μL of a water solution of gold–iron oxide dimers (with an iron concentration ranging from 6 to 12 g L−1) were placed in a sample holder and a fluoro-optic thermometer fibre probe (Luxtron Corp., CA) was used to monitor the temperature for every 0.25 s. The device is capable of generating AC magnetic fields at different frequencies (110 kHz, 220 kHz and 300 kHz) with magnetic field amplitudes up to 24 kA m−1 (for 220 kHz and 300 kHz) and 32 kA m−1 (for 110 kHz). Reported SAR values and error bars were calculated from the mean and standard deviation, respectively, of at least four experimental measurements according to the following equation:where C is the specific heat capacity of water (4185 J L−1 K−1) per unit volume and m is the concentration (g L−1 of Fe) of magnetic material in solution. The measurements were carried out under non-adiabatic conditions, thus the slope of the curve dT/dt was measured by taking into account only the first few seconds of the T vs. time curve (a typical example of the T vs. time profile is reported in the ESI†).
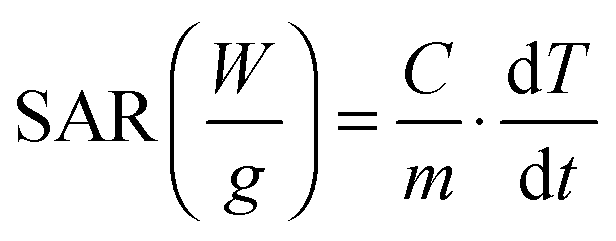
Results and discussion
Two-pot approach
In our two-pot approach, in a surfactant mixture of oleic acid and oleylamine in 1-octadecene (ODE), a solution of pre-made Au NPs (i.e. 9 nm in 1-octadecene, Fig. S1, ESI†) was added, and the mixture was heated following the heating ramp reported in Fig. 1A. Upon injection at 150 °C of Fe(CO)5, and after annealing the solution for 90 min at 200 °C, patchy spots of iron oxide started to nucleate on top of the Au seeds (Fig. 1B). Instead, after aging the solution for 1 h at 300 °C, dumbbell-like dimers were formed, with iron oxide NPs having an average size of 24 nm (Fig. 1C). In this two-pot protocol, by increasing the Fe precursor amount, no change in the iron oxide NP size was possible. Instead, the tuning of the iron oxide NP was possible by deliberately adding a chlorine bearing compound in the reaction mixture (Fig. 2). This is in agreement with other works demonstrating the effect of halide ions on the growth of semiconductor NPs,20–24 and in particular of Cl ions on the growth of metallic iron,25 iron–gold,26 alloyed NPs and on tetrapod-shaped iron oxide NPs.27–30 Here, to tune the iron oxide domain different amounts of a 2 M solution of hydrogen chloride (HCl) in diethyl ether were added to the Au NPs in the reaction flask followed by the injection of the iron precursor. The Cl amount was varied from 0.12 to 0.48 mmol. As a first observation with the addition of chloride ions, dimers were already formed at 200 °C for 90 min (Fig. 2). Moreover rising the amount of Cl ions from 0.12 to 0.24 or to 0.48 mmol led to the tuning of the iron oxide NP domains from 18 to 36 and 42 nm respectively, which are in the range of interest for MH (Fig. 2).31 The nucleation of iron oxide at such a low temperature parallels that observed for gold–metal iron dimers.26 We underline that here, without Cl, dimers were observed only at 300 °C (Fig. 1C). When using 1,2-dichloroethane instead of HCl as a Cl agent, dimers were still formed soon after aging the sample at 200 °C, however larger amounts of the 1,2-dichloroethane were needed to obtain similar iron oxide size domains; at 0.48 mmol or a lower amount of 1,2-dichloroethane, no dimers were found (as instead, this was the case for HCl) and only at 0.625 mmol or 1.25 mmol, dimers having iron oxide NP of 18 nm or 23 nm in size were grown (Fig. S2, ESI†). This difference might be ascribed to the nature and availability of Cl in the chosen compounds; while HCl has an ionic nature, in 1,2-dichloroethane Cl atoms form covalent bonds and they might also evaporate faster during the reaction. | ||
| Fig. 2 Au–FexOy dimers made by a two-pot approach at fixed Fe(CO)5 (0.6 mmol) and increased amounts of HCl: 0.12 (a and b), 0.24 (c and d) and 0.48 mmol (e and f). As shown here, dimers were already formed after 90 min at 200 °C (see the ESI† for details). | ||
One-pot approach
To further simplify the protocol, we tested the in situ formation of Au NPs followed by the growth of iron oxide, in a so-called “one-pot” approach (Fig. 1F). We chose HAuCl4 as a gold precursor since it is the most used Au source and it also provides Cl ions.11 In this case the Au salt (instead of Au NPs) was introduced in the reaction mixture. This could be done either at the very beginning of the reaction, when the reaction mixture was still at room temperature, or after it had been heated at 120 °C, with no significant difference in the outcomes (see Fig. 1D and Fig. S3, ESI†).Similar to the two-pot approach with Cl addition, dimers were formed already at 200 °C as here the Cl ions were provided in the reaction pot as counter-ions of the Au precursor. Size tunability of the iron oxide NPs, in a slightly narrower range (16–26 nm), was achieved by stopping the reaction at 200 °C at fixed times (30, 60 or 90 min, Fig. S4, ESI†).
We also checked the effect of the Fe:Cl ratio on the iron oxide NP size. For instance, in the one-pot approach, when fixing Cl at 0.48 mmol (Cl coming from the Au salt) and increasing Fe(CO)5 from 0.6 mmol (Fe:Cl ratio of 1.25) to 2.25 mmol (Fe:Cl ratio of 4.7), dimers evolved from a dumbbell-like to a flower-like shape after 90 min at 200 °C (Fig. S5A and B, ESI†). If instead the Fe content was fixed at 2.25 mmol and the overall amount of Cl ions was raised from 0.48 to 0.68 mmol (0.48 mmol from the gold salt + 0.2 mmol added as HCl), such that the Fe:Cl ratio was lowered to 3.3, dumbbell-like dimers were formed again (Fig. S5C, ESI†). This suggests that the Fe:Cl ratio controls the size and shape of iron oxide NPs in the dimers. Notably, when using FeCl3·6H2O, as the Fe and Cl source, no dimers were obtained neither at 200 °C nor at 300 °C likely because the ratio Fe:Cl was too low (0.6 mmol of FeCl3 with a Fe:Cl ratio of 0.33) (Fig. S6, ESI†). Finally, attempts to control in situ the size of Au NPs by varying a number of parameters (i.e. gold precursor, surfactant, injection T) were not successful under our conditions (data not shown).
Effects of the 1,2-hexadecanediol (HDDOL) surfactant
We also noted that, in previous reports, dimers with iron oxide NPs of about 20 nm were obtained only if in the synthesis protocol 1,2-hexadecanediol (HDDOL) was not used in the reaction pot.9,22,32–35 This is the reason why in all our syntheses we did not employ HDDOL. However, to verify the effect of the HDDOL on dimers, we performed a parallel control experiment by running a one-pot synthesis in which HDDOL was added while in the other synthesis all the conditions were set the same except for the absence of HDDOL (Fig. S7, ESI†). With HDDOL, amorphous flower-like dimers were formed after the heat treatment at 200 °C for 90 min (Fig. S7E, ESI†). In contrast, dumbbell-like dimers were obtained under the same identical conditions with the only difference that was the absence of HDDOL (Fig. 1D).This suggests that HDDOL hinders the growth of iron oxide. Only a patchy nucleation of the iron oxide on top of Au NPs at 200 °C was seen and an extra step at 300 °C was required to allow the iron oxide domains to coalesce in a single domain (Fig. S7, ESI†). This observation is in agreement with dimers produced by the already reported procedure by Sun and co-workers (Fig. S8 and S9, ESI†). This was also the case for the two-pot method (data not shown).
Characterization of dimers prepared using different methods
Four selected Au–FexOy samples prepared using different syntheses (either one-pot or two-pot) and at different temperatures (either 200 °C or 300 °C) were easily transferred in water using a well-established gallic-polyethylene glycol ligand exchange protocol (see Fig. S10 and S11, ESI†).19 Note that, given the difference in the preparation method, the final dimers compared here differ in the size of the iron oxide NP. Regardless of the route, the best SAR values correspond to dimers having similar iron oxide NP sizes of about 24 and 26 nm in diameter (by two-pot 300 and one-pot 200 respectively).Magnetic and hyperthermia characterizations were performed (Fig. 3 and Fig. S12–S17, ESI†) along with X-ray diffraction (XRD) characterization (Fig. S18, ESI†). The latter confirms the presence of Au and iron oxide phases in all samples. With no possibility to discriminate accurately between γ-Fe2O3/Fe3O4 using XRD, on dimers produced at 200 °C for 90 minutes, using the fastest protocol, we then performed Mössbauer analysis to gain compositional information. Indeed, we could estimate proportions of 46 ± 2% and 54 ± 2% for maghemite and magnetite, respectively (Fig. S20, ESI†). For the sake of simplicity we then labelled the dimers as Au–FexOy. A summary of the sizes along with saturation magnetization (MS) and coercive field (Hc) values at 5 K are given in Table 1 (see Fig. S12 (ESI†) for the magnetization curves). For what concerns the hyperthermia performance, the two-pot 200 sample did not show any heating, even at the highest frequency and field amplitude (data not shown). At 200 °C indeed the dimers were not formed yet and a patchy growth of amorphous iron oxide domains on top of the Au NPs was observed (Fig. 1B).
| Synthesis | T (°C) | Au seeds | d Au (nm) | d FexOy (nm) | d Au–FexOy (nm) | M S (5 K) (emu g−1) | H c (5 K) (Oe) |
|---|---|---|---|---|---|---|---|
| Standard | 300 | No | 6 ± 4 | 15 ± 3 | 21 ± 4 | 88 ± 3 | 280 ± 8 |
| One-pot 200 | 200 | No | 12 ± 3 | 26 ± 5 | 41 ± 5 | 85 ± 5 | 334 ± 3 |
| One-pot 300 | 300 | No | 13 ± 5 | 39 ± 7 | 54 ± 7 | 87 ± 6 | 550 ± 5 |
| Two-pot 200 | 200 | Yes | 12 ± 3 | 6 ± 2 | 18 ± 3 | 28 ± 5 | 518 ± 10 |
| Two-pot 300 | 300 | Yes | 9 ± 2 | 24 ± 4 | 32 ± 6 | 92 ± 4 | 374 ± 5 |
On the other hand, the two-pot 300 sample, whose synthesis differed from the two-pot 200 dimers by a further heating step at 300 °C for 1 h, had a record SAR value of 1030 ± 30 W gFe−1 (24 kA m−1 at 300 kHz) (Fig. 3A and Fig. S13 and S14, ESI†). Indeed the additional heating step allows the iron oxide to coalesce in one single domain, resulting in well-defined dimers (compare Fig. 1B and C).
This is directly reflected on the magnetic parameters: the two-pot 200 sample had lower TB and MS parameters than the two-pot 300 (see Table 1), which are indicative of a smaller magnetic domain along with lower crystallinity of the iron oxide domains. In comparison, a MS value close to the bulk value of Fe2O336 was found for the two-pot 300 sample (Table 1). The Au–FexOy dimers synthesized with the one-pot 200 approach (Fig. S15, ESI†) had SAR values pretty close to that of the two-pot 300 (1010 ± 20 W gFe−1). Instead, when performing an extra heating step at 300 °C, the current one-pot 300 dimers experienced a 30% drop in their SAR compared to that of the one-pot 200 (710 ± 10 W gFe−1 at 300 kHz, for instance) (Fig. S16, ESI†). One possible explanation is due to the final iron oxide domain size; when using the one-pot 300 method the iron oxide domain is larger than that for the one-pot 200 dimers (39 nm versus 26 nm). Such an increase in the size results in a decrease in the SAR values as the optimum size (between 20–24 nm) is overcome.5,19,31 Further, large ferrimagnetic domains show stronger dipolar magnetic interactions which could lead to partial aggregation of the dimers (this can be seen by the hydrodynamic size measured by DLS, Fig. S10, ESI†) and thus to a decrease in SAR.5,19,37 The Au–FexOy dimers synthesized by the addition of HDDOL9 (here referred to as “standard”, Fig. S8, ESI†) and having a 15 nm iron oxide domain exhibited a rather poor heating efficiency, with SAR values barely reaching 100 W gFe−1 (94 ± 4 W gFe−1 at 300 kHz and 24 kA m−1) (Fig. 3 and Fig. S17, ESI†). In this case even by tuning the amount of iron precursor no control over the size domain of iron oxide NPs was possible (Fig. S9, ESI†). Although this sample had a high MS (Table 1), the presence of HDDOL hindered the growth of the iron oxide to only 15 nm, which is far below the optimal size for MH.
SAR characterization as a function of iron oxide domain size
SAR measurements were also performed on iron-oxide dimers prepared using the same one-pot 200 approach, which differ only in the iron oxide size domains (17, 23 and 26 nm obtained at different annealing times of 30, 60 and 90 min, respectively). For comparison, SAR values were also measured on dimers obtained by the same one-pot 200 synthesis with 90 min of annealing time but adding the gold precursor since the beginning of the reaction. In this case the iron oxide domain is of 21 nm in diameter. As shown in Fig. 3C, SAR values increase with the size of the iron oxide domain reaching the maximum values for dimers with an iron oxide domain of 23 nm. This is valid for all the frequencies tested and it is especially evident at 300 and 110 kHz (and at 24 kA m−1). At 110 and 220 kHz a saturation effect is observed for dimers with an iron oxide domain up to 26 nm. Instead at 330 kHz a drop of the SAR values for such a sample is observed when the size of the iron oxide domain goes from 23 up to 26 nm. It is also worth noting that for gold–iron oxide dimers with an iron oxide domain of 23 nm at 300 kHz a very high SAR value (1330 ± 20 W g−1) was observed; this was even higher than that recorded for 35 nm size iron oxide nanocubes under the same field conditions.19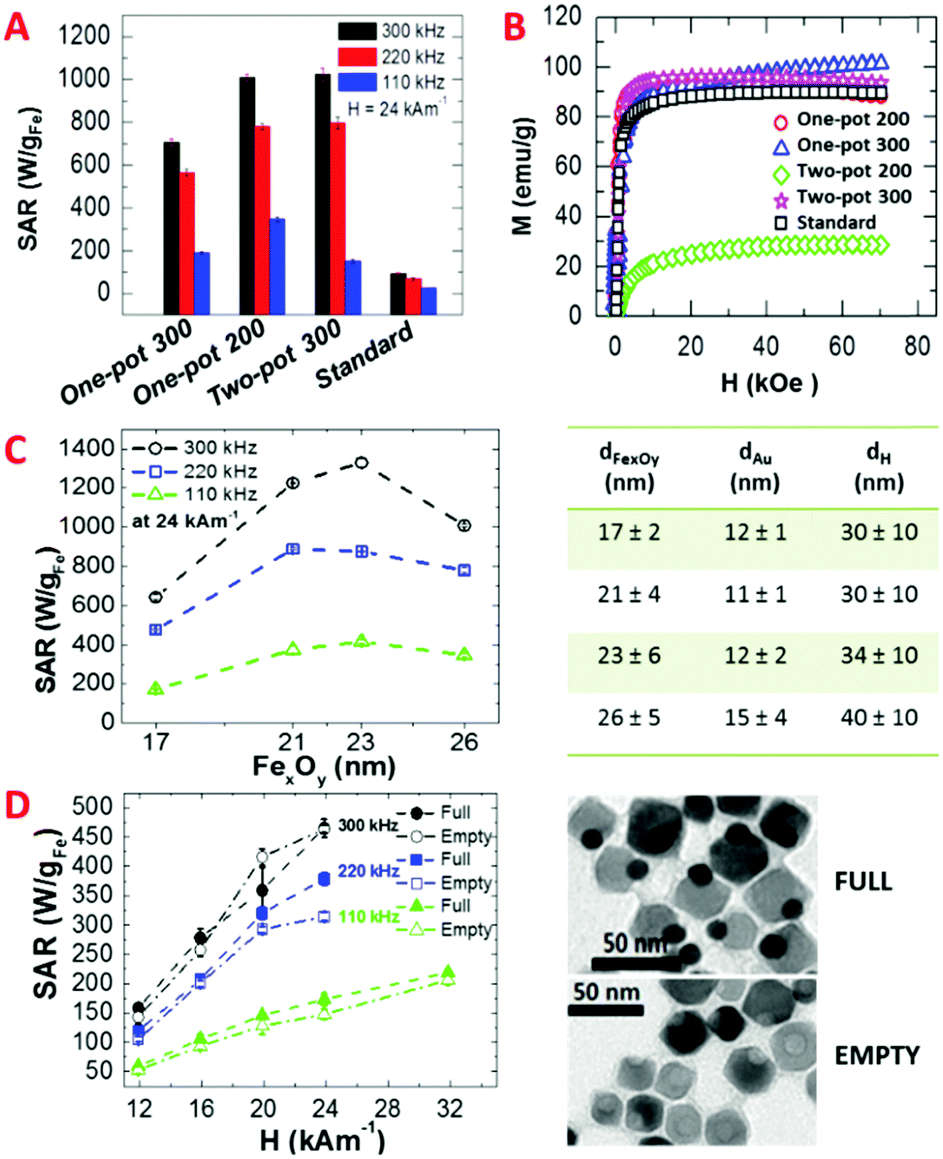 | ||
Fig. 3 (A) SAR values at three frequencies and 24 kA m−1 of Au–FexOy dimers synthesized by different routes. (B) Positive branch of the magnetization vs. magnetic field at 5 K for the dimers made by one-pot 200 (open red circles  ), one-pot 300 (open blue triangles ), one-pot 300 (open blue triangles  ), two-pot 200 (open green rhombi ), two-pot 200 (open green rhombi  ), two-pot 300 (open pink stars ), two-pot 300 (open pink stars  ) and Standard (open black squares □). (C) SAR values at three frequencies and 24 kA m−1 for Au–FexOy dimers with different sizes of the iron oxide domain (from 17 to 26 nm) obtained by one-pot 200 method and different annealing times; in the table the size of the gold and iron oxide domains together with the hydrodynamic size by number, of the measured dimers are reported. (D) Comparative SAR values at three different frequencies for full and empty dimers, the latter obtained by etching out the Au NP: 300 kHz (black full and empty circles ●,○), 220 kHz (blue full and empty squares ) and Standard (open black squares □). (C) SAR values at three frequencies and 24 kA m−1 for Au–FexOy dimers with different sizes of the iron oxide domain (from 17 to 26 nm) obtained by one-pot 200 method and different annealing times; in the table the size of the gold and iron oxide domains together with the hydrodynamic size by number, of the measured dimers are reported. (D) Comparative SAR values at three different frequencies for full and empty dimers, the latter obtained by etching out the Au NP: 300 kHz (black full and empty circles ●,○), 220 kHz (blue full and empty squares  , ,  ) and 110 kHz (green full and empty triangles ) and 110 kHz (green full and empty triangles  , ,  ). Each SAR value is the mean of at least 4 data points. ). Each SAR value is the mean of at least 4 data points. | ||
SAR as a function of the presence of gold
We have also investigated the effect of the gold domain on heat dissipation. On water soluble dimers having a 25 nm iron oxide NP, the gold NP was selectively etched using the Lugol's reagent (an aqueous solution of KI3, see the ESI†), leaving an iron oxide NP with a small cavity on one side. When comparing full dimers with gold-free dimers, it was found that the SAR values of the full dimers were slightly higher than those of the empty dimers, especially at 110 and 220 kHz (Fig. 3C and Fig. S19, ESI†). This suggests that the gold NP might contribute to dissipate the heat produced by the iron oxide NP. Finally, SAR values of both full and empty dimers recorded under patient-safe conditions (Hf < 5 × 109 A m−1 s−1) were in the 600–690 W gFe−1 range, and comparable to those of iron oxide nanocubes that could be considered the benchmark product for MH (Fig. S14–S16, ESI†).19Conclusions
In summary, we have shown that, in the synthesis of Au–FexOy dimers (regardless of whether a one-pot or a two-pot approach was followed) the addition of Cl ions, either as extra ingredients in the reaction pot (i.e. HCl or 1,2-dicholoroethane) or as counter ions of the Au salt (HAuCl4), enables the growth of dimers at relatively low temperatures (200 °C), with a tunable iron oxide domain (above 18 nm) and with a dumbbell-like shape. Exceptional SAR values were reached in dimers with an iron oxide NP of 21–26 nm, with the presence of Au possibly helping the heat dissipation. Both routes are very robust and highly reproducible: the two-pot approach enables the precise tuning of the Au domain by using Au NPs at different sizes, while control over the iron oxide size is achieved by tuning the chloride ion addition. The one-pot synthesis is more cost-effective as it requires less steps and it allows the tuning of the iron oxide size domain by varying the reaction time. In both approaches, by adding chloride ions we could lower the reaction temperature (200 °C rather than 300 °C). Also, we have shown that to grow bigger iron oxide domains, the HDDOL surfactant should be avoided, since it impairs the iron oxide nucleation on top of the gold NPs. Our dimers are promising candidates for in vivo applications offering advanced features compared to bare iron oxide NPs. This is also in light of the early in vivo study showing the long-term biocompatibility and the slow degradation of dimers in one year time in the murine model.38 From the functional point of view, dimers could benefit from the presence of both Au and iron oxide NPs to achieve selective surface attachment on the two different domains (i.e. targeting and drug delivery). Also, in dual modal therapy, when merging MH and photo-ablation, the gold NPs might boost the heat performances of iron oxide NPs with a higher efficiency than that of iron oxide NPs alone.39Acknowledgements
This work is funded by the AIRC project (Contract No. 14527), the European Research Council (starting grant ICARO, Contract No. 678109) and the EU-ITN network Mag(net)icFun (PITN-GA-2012-290248). P. G. thanks the Marie Curie Actions FP7 GA N. 6003888. We thank Dr A. Figuerola for useful discussions.Notes and references
- R. Ivkov, Int. J. Hyperthermia, 2013, 29, 703–705 CrossRef PubMed.
- C. S. S. R. Kumar and F. Mohammad, Adv. Drug Delivery Rev., 2011, 63, 789–808 CrossRef CAS PubMed.
- C. L. Dennis and R. Ivkov, Int. J. Hyperthermia, 2013, 29, 715–729 CrossRef PubMed.
- E. A. Périgo, G. Hemery, O. Sandre, D. Ortega, E. Garaio, F. Plazaola and F. J. Teran, Appl. Phys. Rev., 2015, 2, 041302 Search PubMed.
- P. Guardia, R. Di Corato, L. Lartigue, C. Wilhelm, A. Espinosa, M. Garcia-Hernandez, F. Gazeau, L. Manna and T. Pellegrino, ACS Nano, 2012, 6, 3080–3091 CrossRef CAS PubMed.
- L. Lartigue, D. Alloyeau, J. Kolosnjaj-Tabi, Y. Javed, P. Guardia, A. Riedinger, C. Péchoux, T. Pellegrino, C. Wilhelm and F. Gazeau, ACS Nano, 2013, 7, 3939–3952 CrossRef CAS PubMed.
- J.-H. Lee, J.-T. Jang, J.-S. Choi, S. H. Moon, S.-H. Noh, J.-W. Kim, J.-G. Kim, I.-S. Kim, K. I. Park and J. Cheon, Nat. Nanotechnol., 2011, 6, 418–422 CrossRef CAS PubMed.
- A. Sathya, P. Guardia, R. Brescia, N. Silvestri, G. Pugliese, S. Nitti, L. Manna and T. Pellegrino, Chem. Mater., 2016, 28, 1769–1780 CrossRef CAS.
- H. Yu, M. Chen, P. M. Rice, S. X. Wang, R. L. White and S. Sun, Nano Lett., 2005, 5, 379–382 CrossRef CAS PubMed.
- C. Xu, B. Wang and S. Sun, J. Am. Chem. Soc., 2009, 131, 4216–4217 CrossRef CAS PubMed.
- C. Wang, H. Yin, S. Dai and S. Sun, Chem. Mater., 2010, 22, 3277–3282 CrossRef CAS.
- E. Fantechi, A. G. Roca, B. Sepúlveda, P. Torruella, S. Estrade, F. Peiró, E. Coy, S. Jurga, N. G. Bastús, J. Nogués and V. Puntes, Chem. Mater., 2017, 29, 4022–4035 CrossRef CAS.
- H. Yin, C. Wang, H. Zhu, S. H. Overbury, S. Sun and S. Dai, Chem. Commun., 2008, 4357–4359, 10.1039/b807591c.
- S. Najafishirtari, P. Guardia, A. Scarpellini, M. Prato, S. Marras, L. Manna and M. Colombo, J. Catal., 2016, 338, 115–123 CrossRef CAS.
- C. George, A. Genovese, A. Casu, M. Prato, M. Povia, L. Manna and T. Montanari, Nano Lett., 2013, 13, 752–757 CrossRef CAS PubMed.
- C. Xu, J. Xie, D. Ho, C. Wang, N. Kohler, E. G. Walsh, J. R. Morgan, Y. E. Chin and S. Sun, Angew. Chem., Int. Ed., 2008, 47, 173–176 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- G. Salas, S. Veintemillas-Verdaguer and M. D. P. Morales, Int. J. Hyperthermia, 2013, 29, 768–776 CrossRef PubMed.
- P. Guardia, A. Riedinger, S. Nitti, G. Pugliese, S. Marras, A. Genovese, L. Manna and T. Pellegrino, J. Mater. Chem. B, 2014, 2, 4426–4434 RSC.
- D. Hinrichs, M. Galchenko, T. Kodanek, S. Naskar, N. C. Bigall and D. Dorfs, Small, 2016, 12, 2588–2594 CrossRef CAS PubMed.
- Z. Zhao, Z. Zhou, J. Bao, Z. Wang, J. Hu, X. Chi, K. Ni, R. Wang, X. Chen, Z. Chen and J. Gao, Nat. Commun., 2013, 4, 1–7 CrossRef.
- M. Saruyama, M. Kanehara and T. Teranishi, J. Am. Chem. Soc., 2010, 132, 3280–3282 CrossRef CAS PubMed.
- M. R. Kim, K. Miszta, M. Povia, R. Brescia, S. Christodoulou, M. Prato, S. Marras and L. Manna, ACS Nano, 2012, 6, 11088–11096 CrossRef CAS PubMed.
- T. Guo'an, Z. Jianxin and G. Wanlin, Nanotechnology, 2010, 21, 175601 CrossRef PubMed.
- S. Zhang, G. Jiang, G. T. Filsinger, L. Wu, H. Zhu, J. Lee, Z. Wu and S. Sun, Nanoscale, 2014, 6, 4852–4856 RSC.
- G. Jiang, Y. Huang, S. Zhang, H. Zhu, Z. Wu and S. Sun, Nanoscale, 2016, 8, 17947–17952 RSC.
- A. Shavel, B. Rodríguez-González, M. Spasova, M. Farle and L. M. Liz-Marzán, Adv. Funct. Mater., 2007, 17, 3870–3876 CrossRef CAS.
- A. Shavel, B. Rodríguez-González, J. Pacifico, M. Spasova, M. Farle and L. M. Liz-Marzan, Chem. Mater., 2009, 21, 1326–1332 CrossRef CAS.
- M. V. Kovalenko, M. I. Bodnarchuk, R. T. Lechner, G. Hesser, F. Schäffler and W. Heiss, J. Am. Chem. Soc., 2007, 129, 6352–6353 CrossRef CAS PubMed.
- D. Kim, J. Park, K. An, N.-K. Yang, J.-G. Park and T. Hyeon, J. Am. Chem. Soc., 2007, 129, 5812–5813 CrossRef CAS PubMed.
- B. Mehdaoui, A. Meffre, J. Carrey, S. Lachaize, L.-M. Lacroix, M. Gougeon, B. Chaudret and M. Respaud, Adv. Funct. Mater., 2011, 21, 4573–4581 CrossRef CAS.
- P. Guardia, N. Pérez, A. Labarta and X. Batlle, Langmuir, 2010, 26, 5843–5847 CrossRef CAS PubMed.
- P. Guardia, A. Labarta and X. Batlle, J. Phys. Chem. C, 2011, 115, 390–396 CAS.
- Y. Yu, A. Mendoza-Garcia, B. Ning and S. Sun, Adv. Mater., 2013, 25, 3090–3094 CrossRef CAS PubMed.
- C. Moya, M. D. P. Morales, X. Batlle and A. Labarta, Phys. Chem. Chem. Phys., 2015, 17, 13143–13149 RSC.
- B. D. Cullity, Introduction to Magnetism and Magnetic Materials, Addison-Wesley, Massachusetts, 1972 Search PubMed.
- D. F. Coral, P. Mendoza Zélis, M. Marciello, M. D. P. Morales, A. Craievich, F. H. Sánchez and M. B. Fernández van Raap, Langmuir, 2016, 32, 1201–1213 CrossRef CAS PubMed.
- J. Kolosnjaj-Tabi, Y. Javed, L. Lartigue, J. Volatron, D. Elgrabli, I. Marangon, G. Pugliese, B. Caron, A. Figuerola, N. Luciani, T. Pellegrino, D. Alloyeau and F. Gazeau, ACS Nano, 2015, 9, 7925–7939 CrossRef CAS PubMed.
- A. Espinosa, R. Di Corato, J. Kolosnjaj-Tabi, P. Flaud, T. Pellegrino and C. Wilhelm, ACS Nano, 2016, 10, 2436–2446 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tb00968b |
| ‡ These authors have contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2017 |