
Heteroatom-doped electrodes for all-vanadium redox flow batteries with ultralong lifespan†
Peng
Huang‡
a,
Wei
Ling‡
a,
Hang
Sheng
a,
Yan
Zhou
a,
Xiaopeng
Wu
c,
Xian-Xiang
Zeng
*a,
Xiongwei
Wu
*a and
Yu-Guo
Guo
 *b
*b
aCollege of Science, Hunan Agricultural University, Changsha, Hunan 410128, China. E-mail: wxwcsu05@aliyun.com; xxzeng@hunau.edu.cn
bCAS Key Laboratory of Molecular Nanostructure and Nanotechnology, Institute of Chemistry, Chinese Academy of Sciences (CAS), Beijing 100190, China. E-mail: ygguo@iccas.ac.cn
cAgricultural College, Hunan Agricultural University, Changsha, Hunan 410128, China
First published on 22nd November 2017
Abstract
The phosphorus and fluorine codoped graphite felt electrodes with prominent hydrophilicity present excellent electroactivity towards V2+/V3+ and VO2+/VO2+, elevate the discharging ability up to 250 mA cm−2 and dramatically extend the energy efficiency of vanadium redox flow batteries towards 1000 cycles with 0.003% reduction per cycle.
Renewable and clean energy (such as wind and solar energy, etc.) is considered as the choice of next-generation energy, whose utilization is hindered by its unstable and intermittent nature.1–4 The all-vanadium redox flow battery (VRFB) is regarded as one of the most promising large-scale energy storage devices, due to its high safety, long cycle life and low cost of maintenance.5–14 However, currently available VRFBs are still impeded by a low energy efficiency (EE) and short lifetime.15–17
Among the VRFB components, the electrode is the dominant factor that constrains VRFB performance.18,19 The carbon-based electrode materials20–23 that function as the reaction zone for the redox reactions of V2+/V3+ and VO2+/VO2+ have been studied extensively. The strategies of increasing electrode electro-activity can be generalized into morphology-changed and morphology-preserved modification. The morphology-changed modifications include the growth of metal and metal oxides (Ir,24 Pt,25 Bi,26 Cu,27 Mn3O4,28,29 PbO2,30 WO3,31 MoO2,32 CeO2 (ref. 33)), carbon nanotubes21,34–36 and graphene,37–40 but their stability and cost should be taken into consideration, in addition to degradation of battery lifetime. Heteroatom-doping precisely avoids the aforementioned drawbacks, usually adopting non-metal elements as the doping choice, and is the main avenue for morphology-preserved modification. Much progress based on nitrogen,41,42 chalcogen43 and halogen44 doping has been achieved in previous works. However, the battery durability is barely satisfactory. Consequently, it is still of urgency to explore an ultra-stable electrode with high efficiency.
Herein, the phosphorus and fluorine co-doped graphite felt (PF-GF) electrode was developed via dipping and high-temperature carbonization, and applied in the VRFB. Compared with the pristine GF electrode based VRFB, the VRFB based on the PF-GF electrode operates well at a high current density (250 mA cm−2) and exhibits an EE of 79% after 1000 cycles at 120 mA cm−2 with a 0.003% reduction per cycle. The structural integrity of the PF-GF electrode is preserved after such a long-time circulation. The outstanding performances demonstrate that P and F co-doping results in high-efficiency and can promote the stability of the VRFB.
The PF-GF electrode is acquired through pyrolysis of GF deposited with KPF6; the preparation procedure is schematically illustrated in Fig. 1a. The hydrophilicity was dramatically improved after P and F co-doping compared with that of the pristine GF (Fig. 1a), and no morphology change was observed in the PF-GF from the scanning electron microscopy (SEM) images (Fig. 1c–f) and transmission electron microscopy (TEM) images (Fig. S1a–c†).
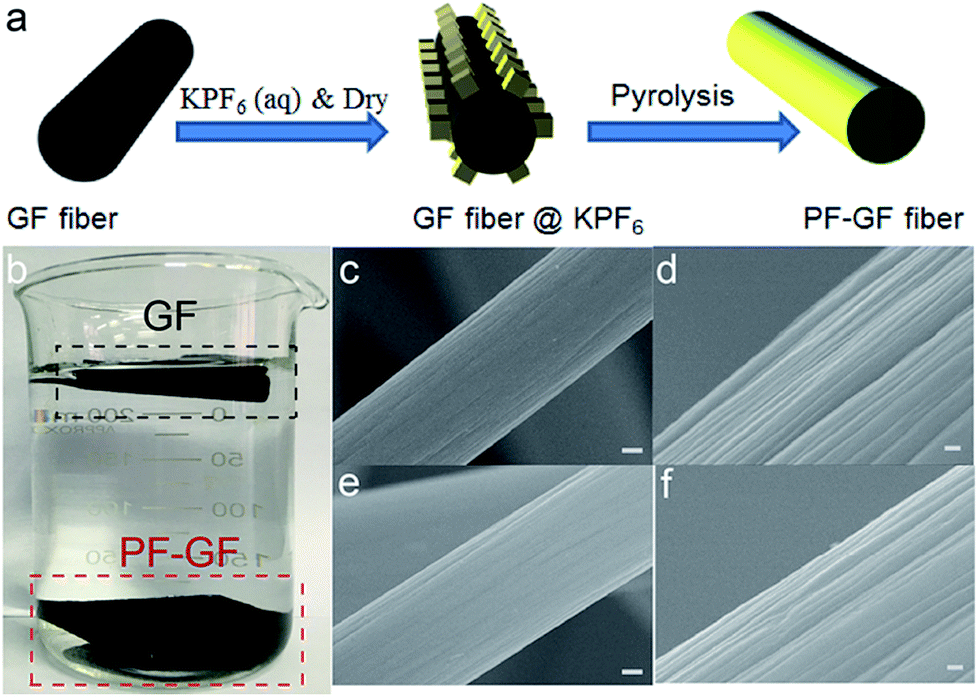 | ||
| Fig. 1 (a) Scheme illustration for preparing the PF-GF electrode. (b) Photograph showing the hydrophilicity of PF-GF and GF. SEM images of GF (c and d) and PF-GF (e and f) with different magnifications. The scale bars in (c) and (e) are 1 μm and the scale bars in (d) and (f) are 100 nm. | ||
To further gain understanding of the chemical composition and functional groups, X-ray photoelectron spectroscopy (XPS) was carried out, and the C 1s and O 1s peaks appear at 284.2 and 532 eV (Fig. 2a), respectively. The O/C ratio of the PF-GF is 0.36, which is higher than that of the GF (0.19) (Fig. S2†). The C 1s spectrum could be deconvoluted into C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (286.5 eV), C–O (286.2 eV) and CC (284.3 eV)45 peaks (Fig. 2b), and F and P signals are also detected in the PF-GF spectrum (Fig. 2c and d). The results possibly originate from the pinning effects of phosphorus and fluorine elements.46 The electro-catalytic effects of P/F atoms can also appear in the carbon materials for other batteries.47,48 Oxygen-containing groups offer more active sites for vanadium ion redox reactions.38 Additionally, the high oxygen content is an explanation of the good hydrophilicity of the PF-GF.
O (286.5 eV), C–O (286.2 eV) and CC (284.3 eV)45 peaks (Fig. 2b), and F and P signals are also detected in the PF-GF spectrum (Fig. 2c and d). The results possibly originate from the pinning effects of phosphorus and fluorine elements.46 The electro-catalytic effects of P/F atoms can also appear in the carbon materials for other batteries.47,48 Oxygen-containing groups offer more active sites for vanadium ion redox reactions.38 Additionally, the high oxygen content is an explanation of the good hydrophilicity of the PF-GF.
 | ||
| Fig. 2 (a) XPS spectrum of the PF-GF and (b) C 1s, (c) F 1s and (d) P 2p spectra. | ||
The P and F bring in plentiful defects. From the Raman spectra (Fig. 3), the intensity ratio of the D and G bands, which represents the degree of graphitization, increases from 0.82 in the GF to 0.99 in the PF-GF. This implies an increment in the number of defects, facilitating many more active sites.
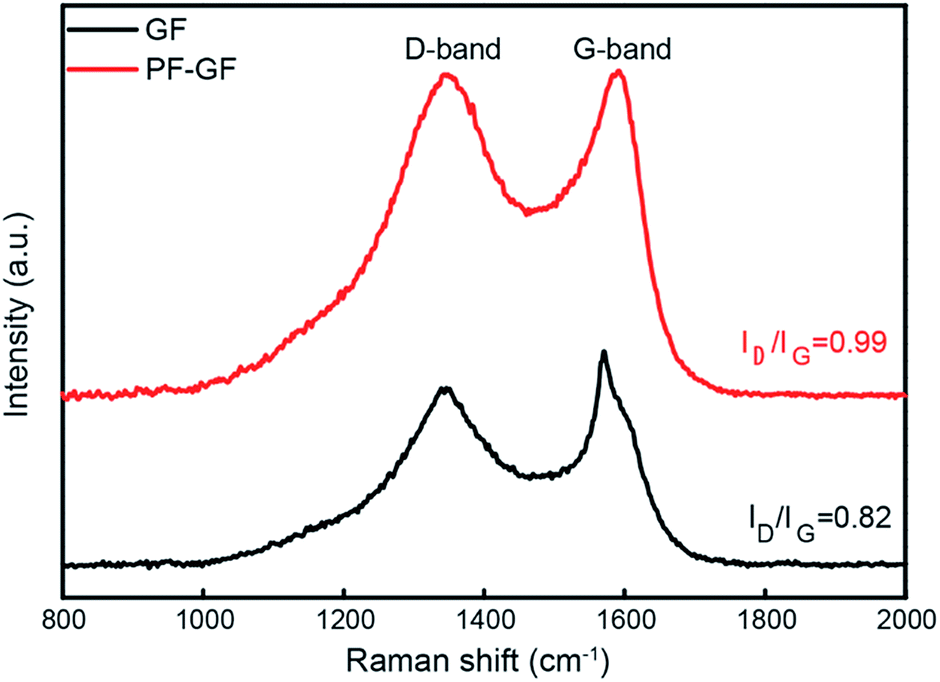 | ||
| Fig. 3 Raman spectra of the PF-GF (red) and GF (black). | ||
The increased number of oxygen-containing groups and defects caused by P and F dramatically promote the reaction of VO2+/VO2+ and V2+/V3+. The cyclic voltammetry curves of the PF-GF electrode show obvious redox reactions. The cathodic and anodic peak potential differences (ΔE) are 457 and 316 mV, respectively (Fig. 4a and b). However, only one peak for the VO2+/VO2+ reaction couple was observed for the GF, and the ΔE is 619 mV. The related parameters are listed in Table 1 and can be found in the ESI.† Meanwhile, the reduction (Ipc) and oxidation (Ipa) peak current densities of the PF-GF electrode are improved, and its absolute value of Ipc/Ipa is closer to 1 for the VO2+/VO2+ couple at various scanning rates (Fig. S3a–d†), which coincides with the ΔE results. In addition, the peak current value density of the PF-GF rises vastly compared with that of the GF. According to the Randles–Sevcik equation (ip = 2.99 × 105α1/2AFnD1/2ν1/2C), the diffusion coefficient (D) of the PF-GF is better than that of the GF, especially for V3+/V2+ (Fig. 4c). The electrochemical impedance spectra of the PF-GF and GF include a semi-circle at the high-frequency region and an oblique line at the low-frequency range, which suggests that the vanadium redox reactions were dominated by charge transfer and diffusion processes. The tremendous reduction of charge transfer resistance demonstrates more favourable electron transfer for the redox reaction (Fig. 4d). The aforementioned results point out that the PF-GF with high reversibility accelerates the redox reaction kinetics.
 | ||
| Fig. 4 The CV profiles of (a) VO2+/VO2+ and (b) V3+/V2+ redox reactions with the PF-GF and GF electrodes at 10 mV s−1. (c) The plots of peak current density versus the square root of the scanning rate for the VO2+/VO2+ redox couples. (d) Impedance tests of the PF-GF and GF electrode in the 0.05 M VOSO4 + 3 M H2SO4 electrolyte with 5 mV amplitude. | ||
| I pa (mA cm−2) | I pc (mA cm−2) | E pa (mV) | E pc (mV) | −Ipc/Ipa | ΔE (mV) | |
|---|---|---|---|---|---|---|
| GF | 88.7 | −52.2 | 1198 | 579 | 0.59 | 619 |
| PF-GF | 106.4 | −89.8 | 1125 | 668 | 0.84 | 457 |
The charging/discharging capabilities of the VRFB based on the PF-GF and GF electrodes were examined in the range from 50 mA cm−2 to 250 mA cm−2. The GF electrode based VRFB fails above 150 mA cm−2. However, the PF-GF electrode has both enhanced energy and voltage efficiency up to 250 mA cm−2 of 54.9% and 56.1% (Fig. 5a and b), respectively. Compared with that of the GF based VRFB, the overpotential of the PF-GF based VRFB reduces by 94 mV, and the discharging capacity increases by 32% from 14.67 to 19.34 Ah L−1 at 100 mA cm−2 (Fig. 5c). During the overall charging/discharging process, the overpotential increases with the increment of current density (Fig. 5d). The increase in overpotential of the PF-GF is not as steep, signifying a lower resistance in the VRFB. The results reveal that the PF-GF electrode with enhanced hydrophilicity has vastly reduced interface resistance with the electrolyte, and adds sites with high electroactivity to accelerate vanadium ion redox reactions.
 | ||
| Fig. 5 The (a) VE and (b) EE of the PF-GF and GF electrodes at various current densities, (c) galvanostatic charging/discharging curves of the PF-GF and GF electrodes at 100 mA cm−2, (d) the plot of overpotential versus current density, and (e) the cycling performance of the PF-GF and GF electrodes at 120 mA cm−2. | ||
After 1000 cycles, the heteroatom doping with oxygen and fluorine plays the main role in stabilizing the electrode, and no morphology change has been observed (Fig. S4 and S5†). As a result, the heteroatom co-doped electrode enables the VRFB to operate well for 1000 cycles at 120 mA cm−2, and the average EE of the cell when using the heteroatom co-doped electrode is 79.2% with 0.003% recession per cycle with slow capacity decay (Fig. 5d and S6†), which indicates the remarkable stability. The heteroatom co-doped electrode displays much better stability and rate capability than GF or other modified GF electrodes in many previous works (Table S1†).
In conclusion, the heteroatom-doped electrode, which acquires P and F from a single dopant, has been demonstrated, and exhibits high electrochemical reversibility and excellent electrocatalytic activity, especially for the V2+/V3+ redox reaction. The VRFB with the heteroatom co-doped electrode reveals splendid rate capability, improved energy and voltage efficiency, and cycling stability. This feasible electrode treatment strategy will inevitably push the VRFB field in a more promising direction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministry of Science and Technology of the People’s Republic of China (Grant No. 2016YFA0202500) and the National Natural Science Foundation of China (Grant No. 51772093).References
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- F. Wang, X. Wu, C. Li, Y. Zhu, L. Fu, Y. Wu and X. Liu, Energy Environ. Sci., 2016, 9, 3570–3611 CAS.
- F. Li and M. Liu, J. Mater. Chem. A, 2017, 5, 15447–15459 CAS.
- N. Xu, X. Li, X. Zhao, J. B. Goodenough and K. Huang, Energy Environ. Sci., 2011, 4, 4942–4946 CAS.
- J. Liu, J.-G. Zhang, Z. Yang, J. P. Lemmon, C. Imhoff, G. L. Graff, L. Li, J. Hu, C. Wang, J. Xiao, G. Xia, V. V. Viswanathan, S. Baskaran, V. Sprenkle, X. Li, Y. Shao and B. Schwenzer, Adv. Funct. Mater., 2013, 23, 929–946 CrossRef CAS.
- Z. Zhang, Z. Cui, L. Qiao, J. Guan, H. Xu, X. Wang, P. Hu, H. Du, S. Li, X. Zhou, S. Dong, Z. Liu, G. Cui and L. Chen, Adv. Energy Mater., 2017, 7, 1602055 CrossRef.
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- K. Wang, K. Jiang, B. Chung, T. Ouchi, P. J. Burke, D. A. Boysen, D. J. Bradwell, H. Kim, U. Muecke and D. R. Sadoway, Nature, 2014, 514, 348–350 CrossRef CAS PubMed.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-González and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 CAS.
- Y.-G. Guo, J.-S. Hu and L.-J. Wan, Adv. Mater., 2008, 20, 2878–2887 CrossRef CAS.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- D. Liu, Z. Wei, Y. Shen, S. D. Sajjad, Y. Hao and F. Liu, J. Mater. Chem. A, 2015, 3, 20322–20329 CAS.
- J. Jiang, Y. Li, J. Liu, X. Huang, C. Yuan and X. W. Lou, Adv. Mater., 2012, 24, 5166–5180 CrossRef CAS PubMed.
- E. SUM and M. Skyllas-Kazacos, J. Power Sources, 1985, 16, 85–95 CrossRef CAS.
- E. SUM and M. Skyllas-Kazacos, J. Power Sources, 1985, 15, 179–190 CrossRef CAS.
- L. Li, S. Kim, W. Wang, M. Vijayakumar, Z. Nie, B. Chen, J. Zhang, G. Xia, J. Hu, G. Graff, J. Liu and Z. Yang, Adv. Energy Mater., 2011, 1, 394–400 CrossRef CAS.
- K. J. Kim, M.-S. Park, Y.-J. Kim, J. H. Kim, S. X. Dou and M. Skyllas-Kazacos, J. Mater. Chem. A, 2015, 3, 16913–16933 CAS.
- A. M. Pezeshki, R. L. Sacci, G. M. Veith, T. A. Zawodzinski and M. M. Mench, J. Electrochem. Soc., 2015, 163, A5202–A5210 CrossRef.
- T. Liu, X. Li, H. Nie, C. Xu and H. Zhang, J. Power Sources, 2015, 286, 73–81 CrossRef CAS.
- M. Park, Y. J. Jung, J. Kim, H. Lee and J. Cho, Nano Lett., 2013, 13, 4833–4839 CrossRef CAS PubMed.
- H. Zhou, Y. Shen, J. Xi, X. Qiu and L. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 15369–15378 CAS.
- X. L. Zhou, T. S. Zhao, Y. K. Zeng, L. An and L. Wei, J. Power Sources, 2016, 329, 247–254 CrossRef CAS.
- S. Chandrabose Raghu, M. Ulaganathan, T. M. Lim and M. Skyllas Kazacos, J. Power Sources, 2013, 238, 103–108 CrossRef CAS.
- R. H. Huang, C. H. Sun, T. m. Tseng, W. k. Chao, K. L. Hsueh and F. S. Shieu, J. Electrochem. Soc., 2012, 159, A1579–A1586 CrossRef CAS.
- B. Li, M. Gu, Z. Nie, Y. Shao, Q. Luo, X. Wei, X. Li, J. Xiao, C. Wang, V. Sprenkle and W. Wang, Nano Lett., 2013, 13, 1330–1335 CrossRef CAS PubMed.
- C. Yao, H. Zhang, T. Liu, X. Li and Z. Liu, J. Power Sources, 2012, 218, 455–461 CrossRef CAS.
- K. J. Kim, M. S. Park, J. H. Kim, U. Hwang, N. J. Lee, G. Jeong and Y. J. Kim, Chem. Commun., 2012, 48, 5455–5457 RSC.
- Y. Ji, J. L. Li and S. F. Y. Li, J. Mater. Chem. A, 2017, 5, 15154–15166 CAS.
- X. Wu, H. Xu, L. Lu, H. Zhao, J. Fu, Y. Shen, P. Xu and Y. Dong, J. Power Sources, 2014, 250, 274–278 CrossRef CAS.
- D. M. Kabtamu, J.-Y. Chen, Y.-C. Chang and C.-H. Wang, J. Mater. Chem. A, 2016, 4, 11472–11480 CAS.
- H. T. Thu Pham, C. Jo, J. Lee and Y. Kwon, RSC Adv., 2016, 6, 17574–17582 RSC.
- H. Zhou, J. Xi, Z. Li, Z. Zhang, L. Yu, L. Liu, X. Qiu and L. Chen, RSC Adv., 2014, 4, 61912–61918 RSC.
- P. Han, Y. Yue, Z. Liu, W. Xu, L. Zhang, H. Xu, S. Dong and G. Cui, Energy Environ. Sci., 2011, 4, 4710–4717 CAS.
- G. Wei, C. Jia, J. Liu and C. Yan, J. Power Sources, 2012, 220, 185–192 CrossRef CAS.
- W. Li, J. Liu and C. Yan, J. Solid State Electrochem., 2013, 17, 1369–1376 CrossRef CAS.
- Z. González, C. Botas, P. Álvarez, S. Roldán, C. Blanco, R. Santamaría, M. Granda and R. Menéndez, Carbon, 2012, 50, 828–834 CrossRef.
- Q. Deng, P. Huang, W.-X. Zhou, Q. Ma, N. Zhou, H. Xie, W. Ling, C.-J. Zhou, Y.-X. Yin, X.-W. Wu, X.-Y. Lu and Y.-G. Guo, Adv. Energy Mater., 2017, 1700461 CrossRef.
- M. Park, I.-Y. Jeon, J. Ryu, J.-B. Baek and J. Cho, Adv. Energy Mater., 2015, 5, 1401550 CrossRef.
- P. Han, H. Wang, Z. Liu, X. Chen, W. Ma, J. Yao, Y. Zhu and G. Cui, Carbon, 2011, 49, 693–700 CrossRef CAS.
- C. Flox, J. Rubio-García, M. Skoumal, T. Andreu and J. R. Morante, Carbon, 2013, 60, 280–288 CrossRef CAS.
- S. Park and H. Kim, J. Mater. Chem. A, 2015, 3, 12276–12283 CAS.
- C. Li, B. Xie, J. Chen, J. He and Z. He, RSC Adv., 2017, 7, 13184–13190 RSC.
- M. Park, I.-Y. Jeon, J. Ryu, H. Jang, J.-B. Back and J. Cho, Nano Energy, 2016, 26, 233–240 CrossRef CAS.
- A. Bismarck, R. Tahhan, J. Springer, A. Schulz, T. Klapötke, H. Zeil and W. Michaeli, J. Fluorine Chem., 1997, 84, 127–134 CrossRef CAS.
- G.-L. Chai, K. Qiu, M. Qiao, M.-M. Titirici, C. Shang and Z. Guo, Energy Environ. Sci., 2017, 10, 1186–1195 CAS.
- C. Zhang, N. Mahmood, H. Yin, F. Liu and Y. Hou, Adv. Mater., 2013, 25, 4932–4937 CrossRef CAS PubMed.
- X. Sun, Y. Zhang, P. Song, J. Pan, L. Zhuang, W. Xu and W. Xing, ACS Catal., 2013, 3, 1726–1729 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta07358e |
| ‡ The authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2018 |