
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal organic framework derived mesoporous carbon nitrides with a high specific surface area and chromium oxide nanoparticles for CO2 and hydrogen adsorption†
Stalin
Joseph
 a,
Devaraju M.
Kempaiah
*a,
Mercy
Benzigar
a,
Arun V.
Baskar
a,
Siddulu N.
Talapaneni
*a,
Sung Hwa
Jhung
b,
Dae-Hwan
Park
a and
Ajayan
Vinu
*a
a,
Devaraju M.
Kempaiah
*a,
Mercy
Benzigar
a,
Arun V.
Baskar
a,
Siddulu N.
Talapaneni
*a,
Sung Hwa
Jhung
b,
Dae-Hwan
Park
a and
Ajayan
Vinu
*a
aFuture Industries Institute (FII), University of South Australia (UniSA), Mawson Lakes Campus, Mawson Lakes, SA 5095, Australia. E-mail: Devaraju.MurukanahallyKempaiah@unisa.edu.au; siddulunaidu@gmail.com; Ajayan.Vinu@newcastle.edu.au
bDepartment of Chemistry, Green-Nano Materials Research Center, Kyungpook National University, Daegu 41566, Republic of Korea
First published on 20th September 2017
Abstract
In this work, we report a simple and versatile method for the preparation of mesoporous carbon nitrides (MCNs) functionalized with highly dispersed chromium oxide nanoparticles by using a metal organic framework, MIL-100(Cr), as a template and aminoguanidine hydrochloride (AG) as a high nitrogen content single molecular precursor. We are able to synthesise these metal oxide functionalized MCN materials with single step carbonization but without using any toxic template removal process using HF or NaOH. The absence of a washing procedure with toxic acid also allows the incorporation of a large amount of metal oxide particles inside the porous channels of MCNs. The obtained MCN materials exhibit a high specific surface area and a large pore volume. The AG to template ratios are varied to control the amine functional groups and the textural parameters including the specific surface area and pore volume. It is found that the AG to template ratio of 1.5 is the best condition to obtain MCNs with a specific surface area of 1294 m2 g−1, which is the highest value reported so far for MCN-based materials. FT-IR and XPS results reveal that the prepared materials contain free NH2 groups within the CN network which help to anchor metal oxide nanoparticles and provide highly dispersed basic sites. These functionalized MCN materials are also used as adsorbents for CO2 capture. Among the materials studied, the MCN with the highest specific surface area shows the largest CO2 adsorption capacity (16.8 mmol g−1) which is much higher than those of MCN materials prepared from SBA-15 and KIT-6, activated carbon, MIL-100(Cr), SBA-15, and multiwalled carbon nanotubes. This high adsorption capacity is mainly due to the strong synergistic effect between the MCN with high specific surface area and highly dispersed metal oxide nanoparticles.
1. Introduction
The rapid growth of energy consumption in the form of fossil fuels creates a lot of problems for the atmosphere due to the enormous CO2 emission.1–5 The as-emitted CO2 can be captured and utilized for several applications such as energy storage, production of chemicals, synthesis of polycarbonate plastics, biological conversion, mineralization and fuel recovery, etc.1 Porous adsorbents such as mesoporous silica, micro and mesoporous carbons and porous carbon nitrides have been widely used as adsorbents for the capture of CO2 owing to their well-ordered porous structures, high specific surface areas and large pore volumes.6–9 Among these adsorbents, mesoporous carbon nitrides (MCNs) are quite unique as they can not only offer well-ordered porous structures and semiconducting properties but also provide basic adsorption sites that are in the form of NH or NH2 groups.10–12 These basic sites help to enhance the adsorption of CO2 molecules which are slightly acidic in nature through a simple acid–base interaction.10,11MCNs are generally synthesized by the nanohard templating approach using mesoporous silicas as templates.12 The properties of the MCN materials can be controlled with a simple adjustment of the structure and pore diameter of the templates and the nature of the nitrogen precursors. With this approach, several MCNs with a large surface area, tuneable pore diameters, and different nitrogen contents have been prepared and used for various other applications including catalysis, sensing, and energy storage and conversion.13–17 For example, since the first report of MCNs by Vinu et al. in 2005,18 various templating approaches such as hard nanocasting and soft self-assembly have been employed for the synthesis of different MCN materials.18–22 Among them, the nanocasting method was found to be an effective way to prepare highly ordered MCN materials and various porous silicas with different structures such as SBA-15, SBA-16,23 KIT-6, and IBN-4 have been widely used as templates.19–21,24–26 This silica templating process generally includes three main steps: (i) infiltration of precursors into the silica template; (ii) polymerization and cross-linking of the carbon and nitrogen precursors into the MCN material at high temperature; and (iii) removal of the silica template with a toxic washing procedure using HF. However, these traditional porous silica templates are expensive and complicated as they involve highly toxic substances such as HF to remove the silica, which limits their use in multifunctional applications with large scale production. By replacing the inorganic templates with organic templates, various steps including the toxic HF treatment, washing with expensive solvents such as ethanol and long synthesis time could be avoided. This simple and single step approach could remove the hurdle in the path of the commercialization of MCNs and help to produce MCN materials in a safe and cost effective way.
Metal–organic frameworks (MOFs) have been considered as a new kind of porous material and also attracted much attention owing to their high adsorption capacities and tuneable pore diameters which make them suitable for various potential applications such as gas separation, storage, catalysis and drug delivery.27–31 The unlimited combinations of organic and inorganic building blocks have offered enormous flexibility to MOFs with high chemical and thermal stability. Among these MOFs, MIL-100(Cr)32 with a chemical composition of Cr3O(F/OH)(H2O)2[C6H3(CO2)3]2·nH2O has great potential due to its large surface area up to 3340 m2 g−1, high pore volume (1.16 cm3 g−1), accessible microporous windows of ca. 0.5 and 0.9 nm, unsaturated chromium sites, unexpectedly high thermal stability (above 300 °C), and rigid structural integrity even after prolonged immersion in water.33 These exciting properties make it an excellent organic templating agent for the fabrication of mesoporous materials including MCNs. However, most of the research on MIL-100(Cr) focused mainly on the structural functionalization and applications in selective adsorption, separation, storage, and catalysis.34,35 By utilising this unique material as a template, not only mesoporous materials could be generated but also with ultra-fine and highly dispersed metal oxide nanoparticles in the pore channels of the mesoporous materials. However, so far, no research has been conducted on using this material as a template for the preparation of MCNs.
Here we describe a silica free, simple and direct synthesis of MCN materials functionalized with chromium oxide nanoparticles and with a large surface area using MIL-100(Cr) as a hard template via a simple thermo-polymerisation of aminoguanidine as a single molecular C and N precursor. To the best of our knowledge, this is the first report on the preparation of MCN materials using a microporous MOF as a sacrificial template along with the incorporation of chromium oxide onto the surface of the MCN materials. The advantages of using porous MIL-100(Cr) as a template are the presence of nanoscale cavities and an open channel structure which could be beneficial in infiltrating the small CN precursor molecules into its open channels to obtain MCN with a high surface area and large pore volume. The prepared MCN exhibits much higher CO2 adsorption capacity than activated carbon, MWCNTs, mesoporous silica, the parent template MIL-100(Cr) and MCN prepared from the SBA-15 template. This simple but effective method can be utilized for the preparation of various MCN nanostructures from different MOFs.
2. Experimental
2.1 Synthesis of MIL-100(Cr)
For the preparation of MIL-100(Cr), a reflux process was carried out using trimesic acid, metallic chromium, HF, HNO3 and H2O as starting materials. The definite amounts of these precursors (trimesic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cr:HF:HNO3:H2O in a 1:1:0.2:0.5:100 weight ratio) were refluxed at 95 °C for 12 h under atmospheric pressure. After the refluxing duration, materials were recovered through purification by washing with hot distilled water and then dried under vacuum.36–38 The final material was used as the template for the preparation of functionalized MCN.
:Cr:HF:HNO3:H2O in a 1:1:0.2:0.5:100 weight ratio) were refluxed at 95 °C for 12 h under atmospheric pressure. After the refluxing duration, materials were recovered through purification by washing with hot distilled water and then dried under vacuum.36–38 The final material was used as the template for the preparation of functionalized MCN.
2.2 Synthesis of chromium oxide functionalized MCN materials
Chromium oxide functionalized MCN materials were prepared through a hard-templating approach using the MIL-100(Cr) as a template (Scheme 1). In a typical synthesis, 1.5 g of aminoguanidine hydrochloride (AG) dissolved in 1.5 g of water was mixed with 1 g of MIL-100(Cr). The resultant MCN-MIL composite was heated at 100 °C for 6 h and further heated at 160 °C for another 6 h in a programmed oven. Subsequently, the composite was heated at 400 °C in a tubular furnace by flowing nitrogen (100 ml per minute) with a heating rate of 3 °C min−1 for about 5 h to achieve the polymerization and subsequent carbonization. A set of samples were prepared with different amounts of AG using the above procedure. The samples are denoted as Cr-MCN-10-X where X denotes the amount of AG in gram added to the synthesis mixture (1.5, 2.0 and 2.5).24,39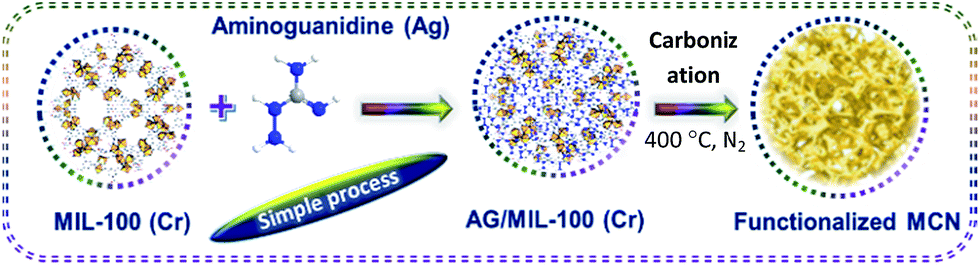 | ||
| Scheme 1 Schematic diagram showing the formation of metal functionalized mesoporous carbon nitride. | ||
2.3 CO2 and H2 adsorption
High pressure CO2 and H2 adsorption measurements were carried out on a Quantachrome Isorb HP1 equipped with a temperature controlled circulator. The CO2 adsorption test was carried out in the pressure range of 0 to 30 bar at 0 to 25 °C after the samples were degassed for 8 h at 200 °C. The H2 adsorption test was carried out at 0 to 50 bar at 50 °C.3. Results and discussion
3.1 Structural analysis of Cr-MCN-10
The structure of the Cr-MCN-10 was analysed by powder X-ray diffraction analysis. Fig. 1A shows the XRD patterns of both the template and Cr-MCN-10-X samples. MIL-100(Cr) shows several peaks at higher angles associated with the cubic crystal system which are in agreement with the previous reports.32 Although the characteristic peaks of the template are clearly visible for MIL-100(Cr), MCN samples prepared with different amounts of AG using MIL-100(Cr) as a template exhibit no peaks in the higher angle associated with the MIL-100(Cr), indicating that the template was completely removed after the carbonization at a high temperature. It is noted that the less intense peaks of the graphitic carbon nitride are observed around (2θ) 25.6° and ca. 44°, which can be attributed to the turbostratic ordering of C and N in the wall structure of the CN framework.12–23 The absence of high intensity peaks at higher angles related to Cr or chromium oxides also confirms that the Cr-based particles are finely dispersed on the pore walls of the Cr-MCN-10 samples and may be amorphous in nature.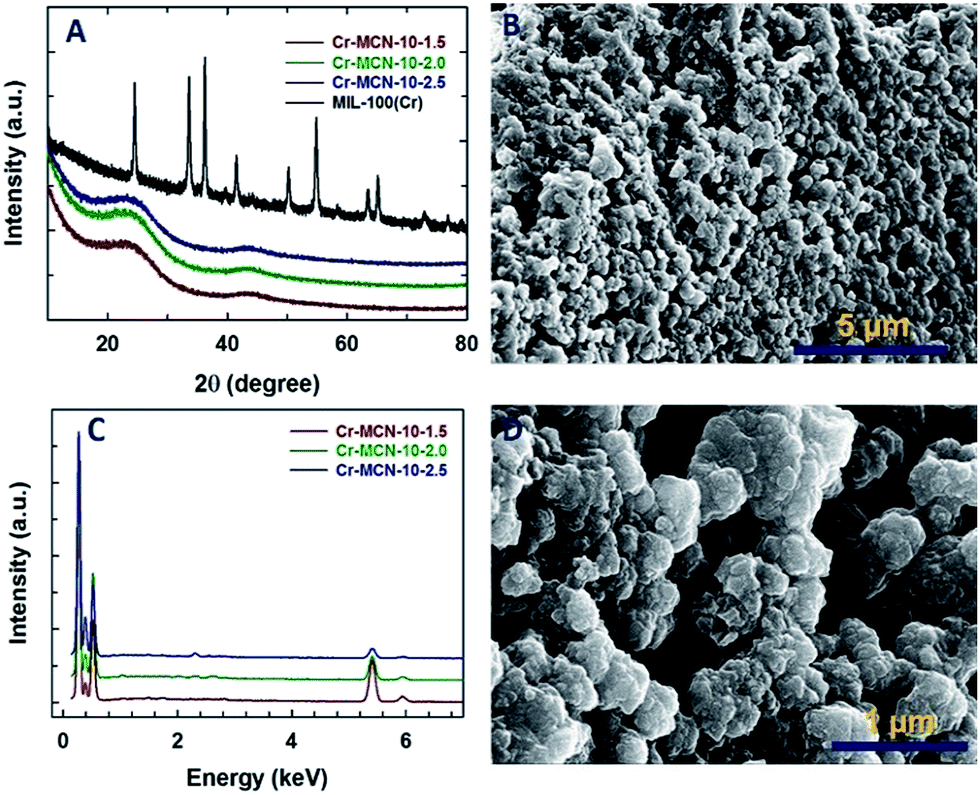 | ||
| Fig. 1 (A) Wide angle XRD patterns of Cr-MCN-10-X and the template MIL-100(Cr), (B) and (D) HRSEM images of Cr-MCN-10-1.5 and (C) energy dispersive X-ray spectroscopy (EDS) patterns of Cr-MCN-10-X. | ||
The HRSEM images of the Cr-MCN-10-1.5 are shown in Fig. 1B and D. The Cr-MCN-10-1.5 possesses highly uniform spherical shaped particles that are highly interconnected and similar to those of the template. The size of the particles is in the range of 100 to 200 nm. It is interesting to note that the macropores are formed between the particles whose sizes are in the range of a few to tens of micrometers. This may be due to the decomposition of the loosely held CN polymers that are decomposed at a high temperature, leaving large mesopores. The energy dispersive X-ray spectroscopy (EDS) pattern of the Cr-MCN-10-1.5 exhibits four sharp peaks which correspond to C, N, O and Cr, revealing that the product is composed of carbon nitride and chromium oxide nanoparticles (Fig. 1C). From the intensity of the Cr peaks, it can be concluded that most of the Cr from the template is transferred to the final Cr-MCN-10 samples as chromium oxide nanoparticles. It is assumed that the Cr atoms reacted with the oxygen of the carboxyl groups in the templates to form the chromium oxide particles. The absence of the chlorine peak in the EDS pattern confirms that the precursor has completely undergone polymerization to form CN polymers and the hydrogen chloride from the precursor is completely removed during the high temperature carbonization process.
It is interesting to note that the intensity of the N peak in the EDS pattern increases concomitantly with the decrease in the intensity of the Cr peak as the amount of AG in the synthesis mixture is increased. This reveals that the content of chromium in the samples prepared with a high amount of AG is lower as compared to that of Cr-MCN-10-1.5. The N/C ratio calculated from the EDS is 0.95–0.46 which is lower than that obtained from the mesoporous silica-based templating procedure (1.33 to 1.9), suggesting that carbon from the template takes part in the reaction.39 This is again confirmed by the fact that the N/C ratio increases with increasing the amount of the precursor in the synthesis mixture. It should also be noted that the particles are agglomerated when the loading of AG is increased in the synthesis mixture (Fig. 1S†). This might be attributed to the formation of CN polymers around the template particles which tend to interact with the neighbouring particles during the polymerization process.
The porosity of the Cr-MCN-10-1.5 is further investigated by high resolution transmission electron microscopy (HRTEM). The HRTEM images reveal that the material is mainly composed of small and slightly disordered mesopores that are quite interconnected and in the size range of 2 to 3 nm (Fig. 2A and B). The elemental mapping of Cr-MCN-10-1.5 further confirms the presence of C, N, O and Cr in the sample. A homogenous distribution of these elements throughout the sample further reveals that the sample is highly uniform and chromium oxide nanoparticles are evenly distributed in the porous channels of the MCN matrix. A similar mesostructure and homogeneous distribution of elements are also observed for other Cr-MCN-10 samples (Fig. 2S to 6S†).
 | ||
| Fig. 2 (A) HRTEM image, (B) bright field scanning TEM image, and (C–F) elemental mapping of Cr-MCN-10-1.5, showing the distribution of carbon, nitrogen chromium and oxygen, respectively. | ||
3.2 Surface area and pore size analysis by nitrogen adsorption
The nitrogen adsorption–desorption isotherms of Cr-MCN-10 materials are shown in Fig. 3A, and the corresponding textural parameters are summarised in Table 1. All the samples show a type IV adsorption isotherm, which is characteristic of porous materials with a mesopore size distribution. The isotherms also show a broad condensation step at lower relative pressure, indicating a small mesopore size. This kind of broad condensation step is a rather peculiar characteristic of materials with bimodal pores or cage like pore systems. These results reveal that the prepared materials have mesopores that indeed originate from the MIL-100(Cr) template due to the decomposition of organic moieties or the displacement of the metal species. There is also a broad hysteresis loop in all of the samples in a higher relative pressure region which is attributed to the large mesopores that originate from the interparticles. It is interesting to note that the amount of nitrogen adsorbed in the monolayer region of the isotherm for Cr-MCN-10 which is used for the calculation of the specific surface area is much higher than those of other MCN samples discovered so far.18,21,23,24,35,39–44 Interestingly, the sample prepared with a low amount of AG registered the highest amount of nitrogen adsorption in the monolayer region than other samples prepared in this study. This indicates that the specific surface area of the samples can be controlled by varying the amount of AG precursor. The textural information of all the samples including the specific surface area, micropore surface area, specific pore volume and pore diameters is shown in Table 1. The specific surface area and the specific pore volume of Cr-MCN-10-1.5 are 1294 m2 g−1 and 0.42 cm3 g−1, respectively. It has been noted that the specific surface area, pore volume and pore diameter of the samples decrease from 1294 to 921 m2 g−1, 0.42 to 0.0.27 cm3 g−1, and 2.10 to 2.0 nm, respectively, as the amount of AG is increased (Table 1). The highest specific surface area of Cr-MCN-10-1.5 mainly originates from the micropores in the sample. This is clearly confirmed by the fact that the micropore area of this sample (771 m2 g−1) is much higher than those of other samples used in this study. However, the specific surface area of the non-porous carbon nitride prepared without the MIL-100(Cr) template is less than 25 m2 g−1. It is interesting to note that all the samples possess bi-modal pores with both micro and mesopores. These pores are formed due to the decomposition of MIL-100(Cr) frameworks during the carbonisation process, in which MIL-100(Cr) behaved as a self-sacrificial template as well as a Cr source for the in situ incorporation of chromium oxides onto the porous surface of Cr-MCN-10. It should be noted that when the amount of AG of 1.0 g was used, the yield of the sample was too low and not enough for the characterization.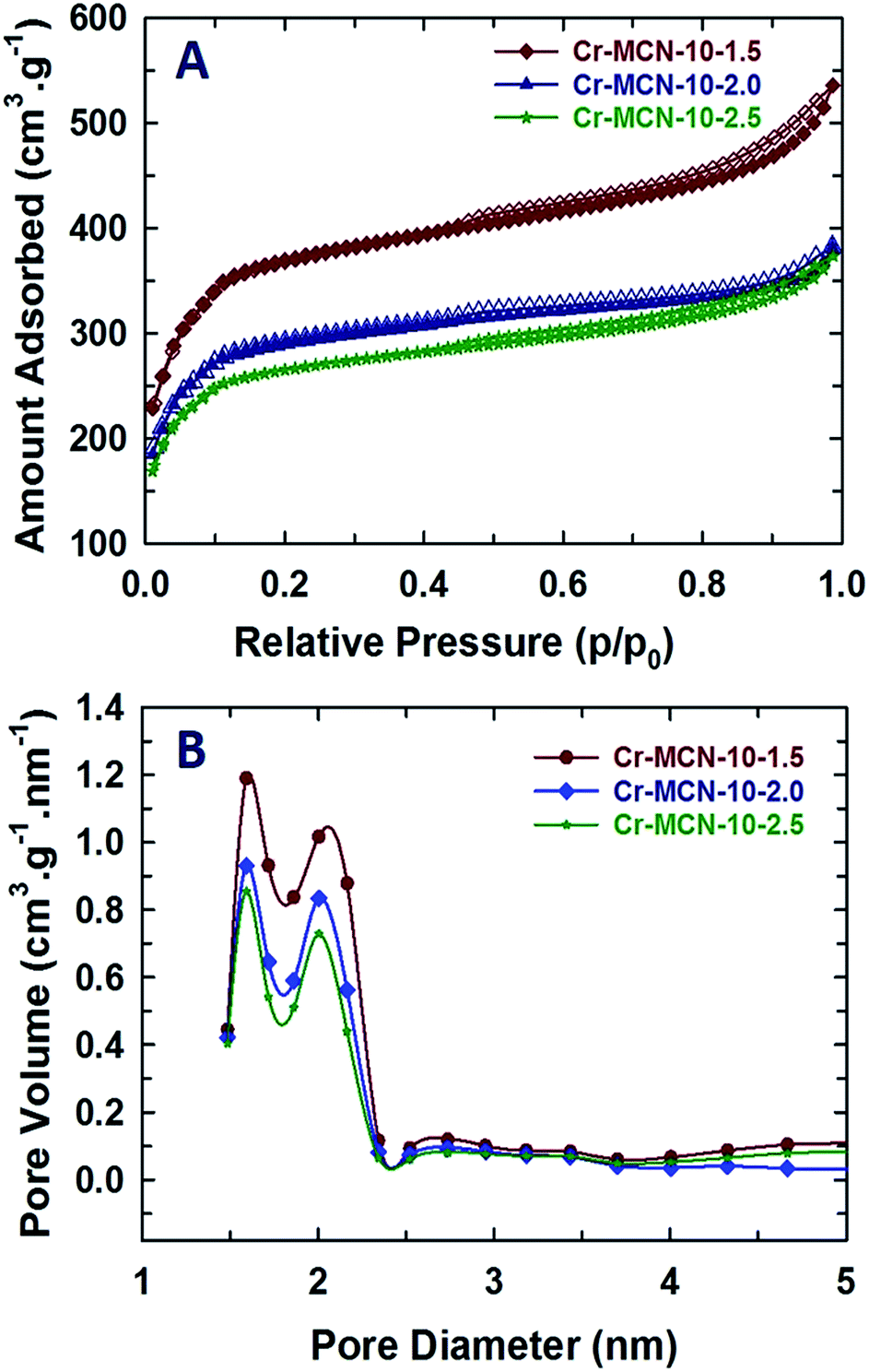 | ||
| Fig. 3 (A) Nitrogen adsorption–desorption isotherms and (B) NLDFT-pore size distributions of Cr-MCN-10-X samples. | ||
| Sample | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Micropore area (m2 g−1) | Pore diameter (nm) | EDS analysis (wt%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| C | N | Cr | O | N/C | |||||
| Cr-MCN-10-1.5 | 1294 | 0.42 | 771 | 1.60, 2.10 | 52.1 | 24.1 | 6.6 | 17.2 | 0.46 |
| Cr-MCN-10-2.0 | 1013 | 0.29 | 641 | 1.57, 2.03 | 53.8 | 25.8 | 3.1 | 17.3 | 0.48 |
| Cr-MCN-10-2.5 | 921 | 0.27 | 583 | 1.57, 2.00 | 42.4 | 40.7 | 0.8 | 16.1 | 0.95 |
3.3 Chemical structure analysis of Cr-MCN-10
Fig. 4A shows the representative electron energy loss spectrum of Cr-MCN-10-1.5. It can be clearly seen that the sample displays sharp C–K, N–K and Cr–L ionization edges that are located at 290.9, 404.2, and 579.4 eV, respectively. The peak at 290.9 eV is attributed to the 1s–π* transition which is caused by the difference in the electron density of C and N due to their different electronegativities. This feature is usually seen for sp2 hybridized carbon in graphite or graphene-based samples and further confirms a strong bonding between the C and N atoms and the C atoms are sp2 hybridised with nitrogen. The peak at 404.2 eV also confirms that the nitrogen atoms are sp2 hybridised with carbon. The sample also shows a higher energy loss which corresponds to the 1s–σ* transition in both C–K and N–K ionization edges, revealing a perfect sp2 hybridization between the C and N atoms. A similar feature was also observed for previously reported MCN-based samples23 but in this case the energy loss for the N–K ionization edges is slightly higher. This might be attributed to the presence of the tetrazine structure in the CN walls of MCN. The Cr-L3 peak appears at 581.1 eV whereas the Cr-L2 split peak is seen at 589.8 eV. These peaks are similar to those observed for chromium oxide nanoparticles and the peak positions are also identical to those observed by Daulton et al.45 A minor and broad peak that is related to oxygen is seen at higher energy which also confirms the presence of chromium oxide nanoparticles in the sample. As previously mentioned, the chromium oxide nanoparticles are formed by the reaction between the chromium atoms and the carboxyl functional groups present in the template.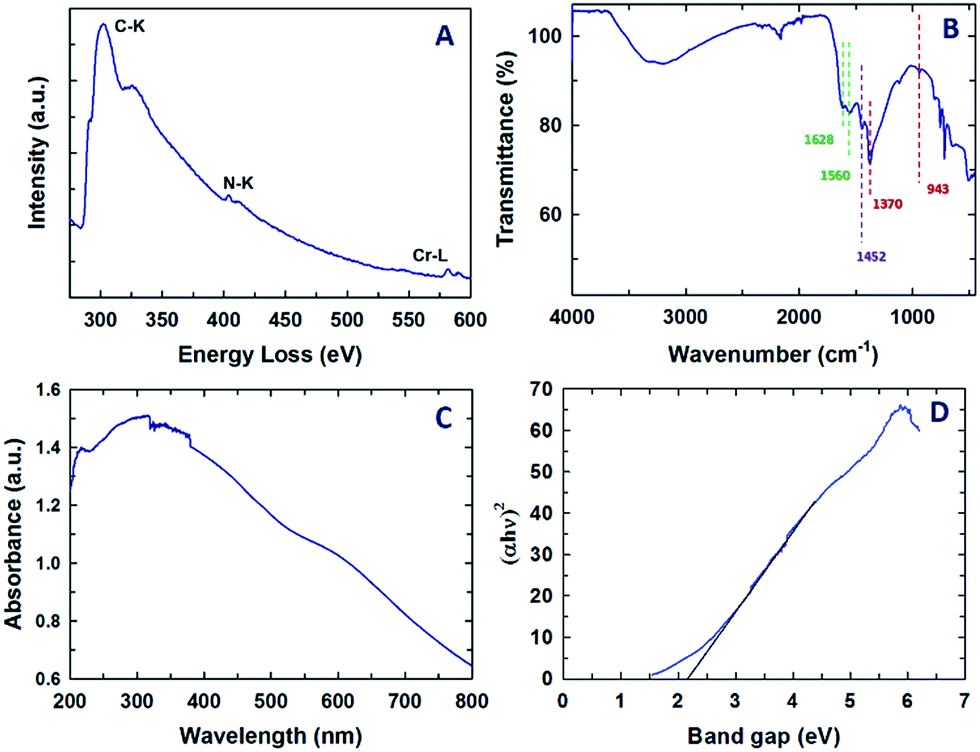 | ||
| Fig. 4 (A) Electron energy loss spectrum, (B) FT-IR spectrum, (C) UV-Vis spectrum and (D) bandgap data of the Cr-MCN-10-1.5 sample. | ||
Information on the chemical bonding between the carbon and nitrogen atoms in the Cr-MCN-10 material was also obtained from FT-IR. The FT-IR spectrum of Cr-MCN-10-1.5 is shown in Fig. 4B. The stretching mode observed in the region 1628 and 1560 cm−1 is assigned to the characteristic vibrations of aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, which is also observed for non-porous CN prepared from AG precursors. The peaks at 943 and 1370 cm−1 also represent the typical tetrazine rings.46 Another band at 1450 cm−1 is assigned to the graphitic C–N band owing to the aromatic ring in the structure.47 The stretching mode between 3150 and 3350 cm−1 is attributed to the N–H stretching of NH2 groups attached to the sp2 hybridized carbon framework, which coincides with the previous reports.12,25 These results obviously support the presence of a CN polymeric network of the carbon nitride structure.
N, which is also observed for non-porous CN prepared from AG precursors. The peaks at 943 and 1370 cm−1 also represent the typical tetrazine rings.46 Another band at 1450 cm−1 is assigned to the graphitic C–N band owing to the aromatic ring in the structure.47 The stretching mode between 3150 and 3350 cm−1 is attributed to the N–H stretching of NH2 groups attached to the sp2 hybridized carbon framework, which coincides with the previous reports.12,25 These results obviously support the presence of a CN polymeric network of the carbon nitride structure.
The UV-visible diffuse reflectance spectrum of Cr-MCN-10-1.5 and its corresponding bandgap data are shown in Fig. 4C and D, respectively. Two broad peaks are observed at 250 and 320 nm and a large broad peak in the visible region (400 to 600 nm) was also observed. The peaks at lower wavelengths are attributed to the π–π* or n–π* transitions in the aromatic CN framework. It is interesting to note that the bandgap of Cr-MCN-10-1.5 is determined to be 2.2 eV from the ultraviolet-differential reflectance spectrum as shown in Fig. 3D. The pure MCN materials have the energy bandgap of 2.7 eV whereas the MCN prepared from AG using mesoporous silica as templates exhibits the bandgap of 2.25–2.49 eV. After incorporation of Cr into the CN structure, the bandgap of the material is decreased from 2.7 to 2.2 eV. The lower bandgap is highly necessary for the photocatalytic application involving water splitting or photocatalytic degradation. The presence of Cr-based nanoparticles will also provide catalytically active redox sites or more basicity to the materials. It has been reported that24 the bandgap of porous carbon nitride materials could be tuned in the range of 2.26 to 2.69 eV with a simple adjustment of the type and nature of the bonding structure and/or the nitrogen contents. It is worth noting that we have successfully prepared the Cr-MCN-10 with a much lower bandgap in the presence of chromium without any doping reagent.
X-ray photoelectron spectroscopy (XPS) analysis was also carried out for the Cr-MCN-10-1.5 and the results are shown in Fig. 5A–D. The XPS survey spectrum shows the peaks related to Cr, C, N and O (Fig. 5A). Interestingly, the intensity of the nitrogen peaks increases with increasing the amount of AG in the synthesis mixture. This indicates that the nitrogen content of the materials increases with the addition of more amount of AG. The presence of the O peak confirms the presence of a trace amount of oxygen which might have originated from the adsorbed CO2 or chromium oxide nanoparticles. This result is consistent with the data obtained from EDS analysis. The chemical bonding between carbon, nitrogen and chromium atoms has been obtained from the high resolution C 1s, N 1s and Cr 2p spectra of Cr-MCN-10-1.5 which are shown in Fig. 5. The deconvoluted C 1s spectrum shows four peaks around 288.1, 286.9, 285.7, and 284.6 eV (Fig. 5B). The large peak at the lowest energy contribution is due to the sp2 hybridized carbon in the sample which is quite different from the sample prepared from mesoporous silica as the template. The peak at 285.7 eV is attributed to the sp2 hybridized carbon bonded to nitrogen inside the aromatic ring whereas the peak at 286.9 eV is ascribed to carbon bonded with nitrogen present in the aromatic tetrazine ring (Scheme S1†).21 The highest energy contribution at 288.1 eV might have originated from the carbon bonded with the trigonal nitrogen atoms.24,42 In the N 1s XPS spectra, we can clearly observe two strong peaks centered at 398.7 and 400.2 eV (Fig. 5C). The lowest energy contribution is assigned to nitrogen atoms bonded with sp2 hybridised carbon whereas the highest energy contribution is attributed to the trigonal nitrogen phase bonded to sp2 hybridised carbon. The observed binding energies of C 1s and N 1s spectra closely matched with the reported binding energies of MCN materials.48,49 The Cr 2p spectrum reveals that the binding energies of Cr 2p3/2 and Cr 2p1/2 fall at 576.5 and 586.6 eV, respectively (Fig. 5D). These peaks are attributed to Cr3+ and Cr6+ in Cr2O3 and CrO3 nanoparticles in MCN. Both the Cr 2p3/2 and Cr 2p1/2 peaks are deconvoluted into two valence states: low-valence (Cr3+) at 576.6 eV and high-valence (Cr6+) at 579.3 eV, respectively, indicating a mixed phase of Cr2O3 and CrO3 nanoparticles.50–52 These results confirm that ultra-fine chromium oxide nanoparticles which are formed on the porous channels of MCN after the decomposition of the MIL-100(Cr) template are present in the form of chromium oxide in Cr-MCN-10.
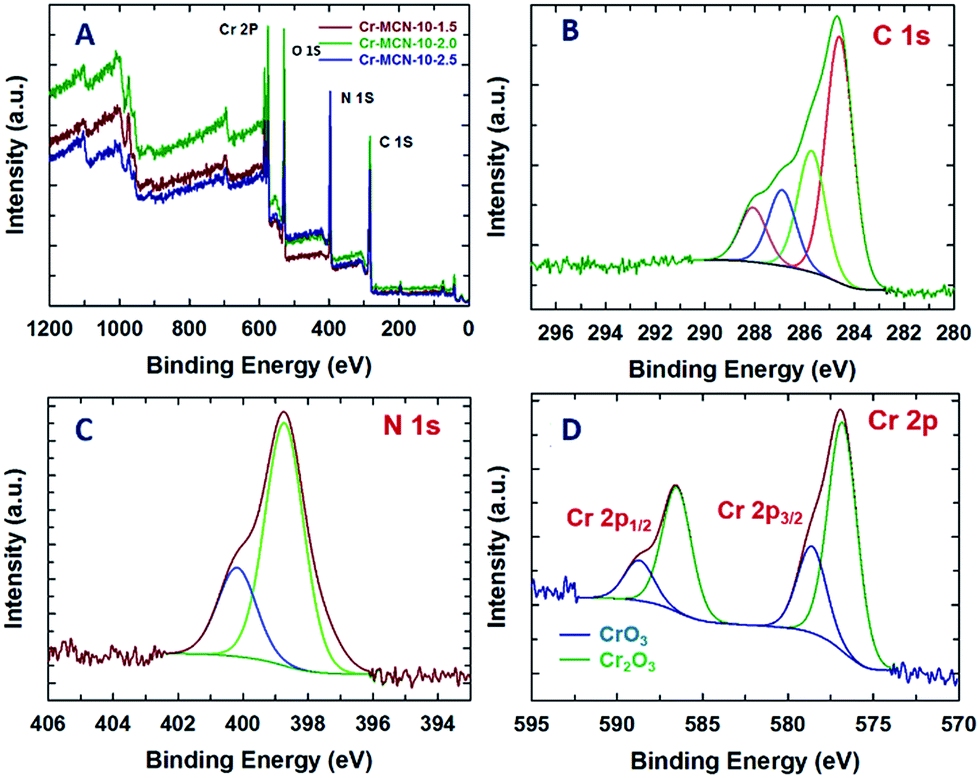 | ||
| Fig. 5 (A) XPS survey spectra of Cr-MCN-10-X samples, (B) deconvoluted C 1s spectra, (C) N 1s spectra and (D) Cr 2p spectra of the Cr-MCN-10-1.5 sample. | ||
3.4 CO2 and H2 adsorption analysis
The presence of a highly basic nitrogen content in Cr-MCN-10-X samples with a large surface area encourages us to evaluate them for the CO2 adsorption. The CO2 capturing ability of Cr-MCN-10-X samples was investigated at high pressures up to 30 bars and at different temperatures and the results are shown in Fig. 6A–D. Fig. 6A shows the CO2 adsorption isotherms of Cr-MCN-10 materials prepared with different amounts of AG precursors. Among the samples studied, Cr-MCN-10-1.5 showed the highest CO2 adsorption capacity of 16.8 mmol g−1 at the adsorption temperature of 0 °C and at 30 bar. This large CO2 adsorption capacity is attributed to the large surface area, micropore area, and nitrogen functionalities on the surface which provide the basic sites. Interestingly, Cr-MCN-10-2.5 exhibits the higher adsorption capacity (10.7 mmol g−1) than Cr-MCN-10-2.0 (10.0 mmol g−1) even though the latter has a higher specific surface area than the former. This is explained by the fact that Cr-MCN-10-2.5 has a higher nitrogen content which provides more basic sites than that of Cr-MCN-10.2.0. However, Cr-MCN-10-1.5 has the lowest nitrogen content but it is still found to be the best adsorbent. It is surmised that the presence of a large amount of chromium oxide nanoparticles in this material provides a synergetic effect with more basicity that enhances the amount of CO2 adsorbed. This result confirms that not only the nitrogen content but also large specific surface area and metal oxide nanoparticles are critical to enhance the CO2 adsorption capacity. It is also found that the amount of CO2 adsorbed can be controlled with a simple adjustment of the chromium oxide nanoparticles in the pore channels of the Cr-MCN-10.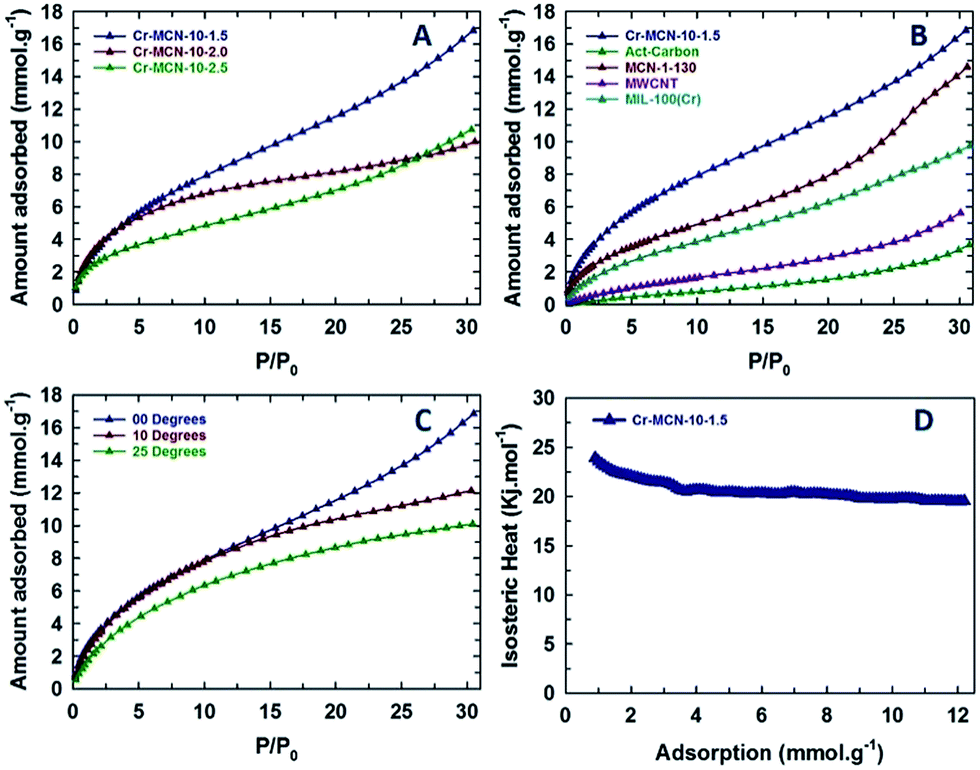 | ||
| Fig. 6 (A) CO2 adsorption isotherms of Cr-MCN-10-X samples at zero degrees, (B) comparison of CO2 adsorption isotherms of the Cr-MCN-10-1.5 sample with other well established porous materials, (C) CO2 adsorption isotherms of the Cr-MCN-10-1.5 sample in different temperature ranges and (D) heat of adsorption plot of the Cr-MCN-10-1.5 sample. | ||
In order to verify the superiority of the Cr-MCN-10 materials for the CO2 adsorption, the CO2 adsorption capacity of Cr-MCN-10-1.5 was compared with those of the template, MIL-100(Cr), activated carbon, multiwalled carbon nanotube and MCN prepared from mesoporous silica templates. It is interesting to note that the CO2 adsorption capacity of Cr-MCN-10-1.5 is much higher than those of the template MIL-100(Cr) (9.8 mmol g−1), activated carbon (3.6 mmol g−1), and other carbon nitride MCN-1-130 (14.5 mmol g−1).6,12,36,37 These results demonstrate the superior nature of the Cr-MCN-10 that was prepared in a single step without any hazardous HF washing procedure for the CO2 adsorption. The effect of adsorption temperature on the CO2 adsorption capacity was also investigated. Fig. 6C shows the CO2 adsorption isotherms of Cr-MCN-10-1.5 measured at different temperatures (0, 10 and 25 °C). As can be seen in Fig. 6C, the amount of CO2 adsorbed increases with increasing the analysis temperature and the adsorption temperature of 0 °C was found to be the best. The nature of the interaction between the adsorbate and the adsorbent was analyzed by calculating the isosteric heat of adsorption using the Clausius–Clapeyron equation. The sample exhibits a heat of adsorption of 25 kJ at low CO2 adsorption to 21 kJ as the amount of CO2 is increased. The value of heat of adsorption at higher CO2 loading is almost similar to those of MCN-1 materials but higher than those of mesoporous carbon-based adsorbents. This clearly explains the role of nitrogen functionalities and chromium oxides in the CO2 adsorption. In addition, the excellent CO2 adsorption performance is the result of weak van der Waals forces and the capillary condensation effect due to the mesoporous and microporous nature of the pores and the chemical bonding with the CO2 and specific functionalities owing to the presence of abundant Lewis base sites in the active catalyst.53
The hydrogen adsorption properties of the prepared Cr-MCN-10 samples were also investigated and the results are compared with those of the template MIL-100(Cr) (Fig. 7). It should be noted that Cr-MCN-10-1.5 registered the hydrogen adsorption capacity of 22.5 mmol g−1 which is much higher than those of other Cr-MCN-10 samples and the template MIL-100(Cr). Again, the higher hydrogen adsorption capacity of this material is attributed to the large surface area and the presence of a small amount of chromium oxide nanoparticles that offer a synergistic effect which is crucial for enhancing the amount of hydrogen adsorption. All these results clearly confirm that the prepared materials are excellent candidates for the adsorption of hydrogen or CO2 molecules and have the potential to replace the existing highly ordered porous materials owing to their ease of preparation and low cost.
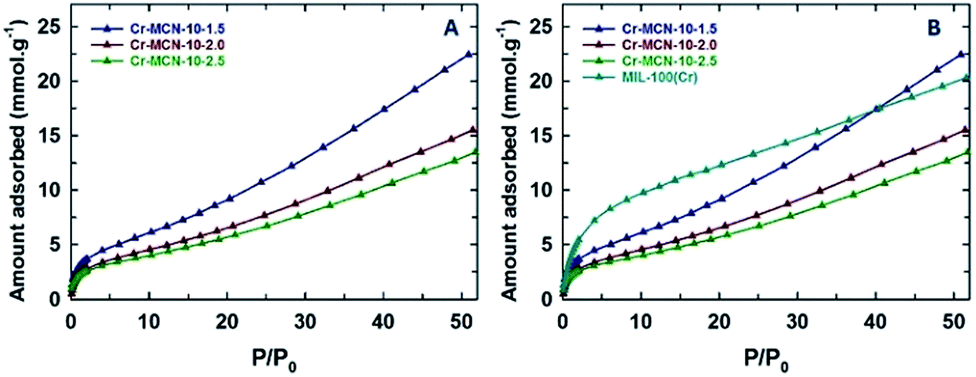 | ||
| Fig. 7 (A) H2 adsorption isotherms of Cr-MCN-10-X samples at zero degrees and (B) comparison of H2 adsorption isotherms of Cr-MCN-10-X samples with the template MIL-100(Cr). | ||
4. Conclusions
In this work, we demonstrated a novel route for a single step synthesis of porous carbon nitrides with ultra-fine metal oxide nanoparticles using MIL-100(Cr) as a sacrificial template without any toxic HF-based template removal process. We demonstrated that MCN materials can be decorated with ultra-fine chromium oxide nanoparticles without any post-synthetic treatment. The prepared mesoporous carbon nitride materials exhibited much higher specific surface areas and pore volumes than the reported mesoporous carbon nitrides prepared from mesoporous silica templates. XPS, EELS and FT-IR results confirmed that the Cr-MCN-10 materials have the typical graphitic CN chemical structure that is composed of tetrazine moieties. We also found that the bandgap of the functionalized Cr-MCN-10 material (2.2 eV) was much lower than those of non-porous MCN and other MCN materials (2.7 eV), which is very beneficial for photocatalytic applications involving water splitting. We also demonstrated that the Cr-MCN-10 samples showed much higher CO2 and hydrogen adsorption capacity than the template, activated carbon, multiwalled carbon nanotubes and porous MCN nanostructures prepared from mesoporous silica templates. We concluded that these exciting adsorption properties are due to the presence of large specific surface area, micropore surface area, a large number of basic nitrogen functionalities and the synergistic effects provided by the ultra-fine chromium oxide nanoparticles. We believe that the approach presented in this work is quite simple and could be used for the preparation of a series of CN materials with different metal oxide nanoparticles using various metal organic framework materials as templates. Moreover, the prepared materials also have the potential to replace the currently available adsorbent materials for carbon capture and hydrogen adsorption and could be used as catalysts for various redox-based catalytic reactions.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
One of the authors A. Vinu acknowledges the Australian Research Council for the Future Fellowship, ARC Discovery Project Awards (DP 150104828; DP 170104478; FT100100970), and University of South Australia for start-up grants.References
- D. Y. C. Leung, G. Caramanna and M. M. Maroto-Valer, Renewable Sustainable Energy Rev., 2014, 39, 426–443 CrossRef CAS.
- T. Seki, Y. Kokubo, S. Ichikawa, T. Suzuki, Y. Kayaki and T. Ikariya, Chem. Commun., 2009, 349–351 RSC.
- R. A. Betts, O. Boucher, M. Collins, P. M. Cox, P. D. Falloon, N. Gedney, D. L. Hemming, C. Huntingford, C. D. Jones, D. M. Sexton and M. J. Webb, Nature, 2007, 448, 1037–1041 CrossRef CAS PubMed.
- J. Meyer, Nature, 2008, 455, 733 CrossRef CAS.
- M. Z. Jacobson, Energy Environ. Sci., 2009, 2, 148–173 CAS.
- L. Liu, F. Y. Wang, G. S. Shao and Z. Y. Yuan, Carbon, 2010, 48, 2089–2099 CrossRef CAS.
- M. Sevilla, P. Valle-Vigon and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781–2787 CrossRef CAS.
- L. Liu, F. Y. Wang, G. S. Shao, T. Y. Ma and Z. Y. Yuan, Carbon, 2010, 48, 2660–2664 CrossRef CAS.
- C. Pevida, T. C. Drage and C. E. Snape, Carbon, 2008, 46, 1464–1474 CrossRef CAS.
- J. L. Zimmerman, R. Williams, V. N. Khabashesku and J. L. Margrave, Nano Lett., 2001, 1, 731–734 CrossRef CAS.
- Q. Li, J. P. Yang, D. Feng, Z. X. Wu, Q. L. Wu, S. S. Park, C. S. Ha and D. Y. Zhao, Nano Res., 2010, 3, 632–642 CrossRef CAS.
- Q. F. Deng, L. Liu, X. Z. Lin, G. H. Du, Y. P. Liu and Z. Y. Yuan, Chem. Eng. J., 2012, 203, 63–70 CrossRef CAS.
- M. Kawaguchi, S. Yagi and H. Enomoto, Carbon, 2004, 42, 345–350 CrossRef CAS.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Muller, R. Schlogl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- Y. Qiu and L. Gao, Chem. Commun., 2003, 2378–2379 RSC.
- E. Kroke and M. Schwarz, Coord. Chem. Rev., 2004, 248, 493–532 CrossRef CAS.
- M. Groenewolt and M. Antonietti, Adv. Mater., 2005, 17, 1789–1792 CrossRef CAS.
- A. Vinu, K. Ariga, T. Mori, T. Nakanishi, S. Hishita, D. Golberg and Y. Bando, Adv. Mater., 2005, 17, 1648–1652 CrossRef CAS.
- Y. S. Jun, W. H. Hong, M. Antonietti and A. Thomas, Adv. Mater., 2009, 21, 4270–4274 CrossRef CAS.
- E. Z. Lee, Y. S. Jun, W. H. Hong, A. Thomas and M. M. Jin, Angew. Chem., Int. Ed., 2010, 49, 9706–9710 CrossRef CAS PubMed.
- A. Vinu, Adv. Funct. Mater., 2008, 18, 816–827 CrossRef CAS.
- S. N. Talapaneni, J. H. Lee, S. H. Je, O. Buyukcakir, T. W. Kwon, K. Polychronopoulou, J. W. Choi and A. Coskun, Adv. Funct. Mater., 2017, 27, 1604658 CrossRef.
- A. Vinu, P. Srinivasu, D. P. Sawant, T. Mori, K. Ariga, J. S. Chang, S. H. Jhung, V. V. Balasubramanian and Y. K. Hwang, Chem. Mater., 2007, 19, 4367–4372 CrossRef CAS.
- S. N. Talapaneni, G. P. Mane, A. Mano, C. Anand, D. S. Dhawale, T. Mori and A. Vinu, ChemSusChem, 2012, 5, 700–708 CrossRef CAS PubMed.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- B. Wang, A. P. Cote, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nature, 2008, 453, 207–211 CrossRef CAS PubMed.
- A. L. Dzubak, L. C. Lin, J. Kim, J. A. Swisher, R. Poloni, S. N. Maximoff, B. Smit and L. Gagliardi, Nat. Chem., 2012, 4, 810–816 CrossRef CAS PubMed.
- A. Torrisi, R. G. Bell and C. Mellot-Draznieks, Cryst. Growth Des., 2010, 10, 2839–2841 CAS.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- D. Marton, K. J. Boyd, A. H. Al-Bayati, S. S. Todorov and J. W. Rabalais, Phys. Rev. Lett., 1994, 73, 118–121 CrossRef CAS PubMed.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC.
- G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surblé, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef PubMed.
- M. Latroche, S. Surble, C. Serre, C. Mellot-Draznieks, P. L. Llewellyn, J. H. Lee, J. S. Chang, S. H. Jhung and G. Ferey, Angew. Chem., Int. Ed., 2006, 45, 8227–8231 CrossRef CAS PubMed.
- Y. Zhong, M. Jaidann, Y. Zhang, G. X. Zhang, H. Liu, M. I. Ionescu, R. Y. Li, X. L. Sun, H. Abou-Rachid and L. S. Lussier, J. Phys. Chem. Solids, 2010, 71, 134–139 CrossRef CAS.
- K. S. Lakhi, W. S. Cha, S. Joseph, B. J. Wood, S. S. Aldeyab, G. Lawrence, J. H. Choy and A. Vinu, Catal. Today, 2015, 243, 209–217 CrossRef CAS.
- M. N. Timofeeva, V. N. Panchenko, N. A. Khan, Z. Hasan, I. P. Prosvirin, S. V. Tsybulya and S. H. Jhung, Appl. Catal., A, 2017, 529, 167–174 CrossRef CAS.
- M. N. Timofeeva, V. N. Panchenko, A. A. Abel, N. A. Khan, I. Ahmed, A. B. Ayupov, K. P. Volcho and S. H. Jhung, J. Catal., 2014, 311, 114–120 CrossRef CAS.
- P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. De Weireld, J. S. Chang, D. Y. Hong, Y. Kyu Hwang, S. Hwa Jhung and G. Ferey, Langmuir, 2008, 24, 7245–7250 CrossRef CAS PubMed.
- S. N. Talapaneni, G. P. Mane, D. Park, K. S. Lakhi, K. Ramadass, S. Joseph, W. M. Skinner, U. Ravon, K. Al-Bahily and A. Vinu, J. Mater. Chem. A, 2017, 5, 18183–18192 CAS.
- X. Jin, V. V. Balasubramanian, S. T. Selvan, D. P. Sawant, M. A. Chari, G. Q. Lu and A. Vinu, Angew. Chem., Int. Ed., 2009, 48, 7884–7887 CrossRef CAS PubMed.
- S. N. Talapaneni, S. Anandan, G. P. Mane, C. Anand, D. S. Dhawale, S. Varghese, A. Mano, T. Mori and A. Vinu, J. Mater. Chem., 2012, 22, 9831–9840 RSC.
- G. P. Mane, D. S. Dhawale, C. Anand, K. Ariga, Q. M. Ji, M. A. Wahab, T. Mori and A. Vinu, J. Mater. Chem. A, 2013, 1, 2913–2920 CAS.
- G. P. Mane, S. N. Talapaneni, K. S. Lakhi, H. Ilbeygi, U. Ravon, K. Al-Bahily, T. Mori, D. H. Park and A. Vinu, Angew. Chem., Int. Ed., 2017, 56, 8481–8485 CrossRef CAS PubMed.
- D. H. Park, K. S. Lakhi, K. Ramadass, M. K. Kim, S. N. Talapaneni, S. Joseph, U. Ravon, K. Al-Bahily and A. Vinu, Chem.–Eur. J., 2017, 23, 10753–10757 CrossRef CAS PubMed.
- T. L. Daulton and B. J. Little, Ultramicroscopy, 2006, 106, 561–573 CrossRef CAS.
- M. Kim, S. Hwang and J. S. Yu, J. Mater. Chem., 2007, 17, 1656–1659 RSC.
- W. J. Ong, L. L. Tan, S. P. Chai and S. T. Yong, Dalton Trans., 2015, 44, 1249–1257 RSC.
- W. J. Son, J. S. Choi and W. S. Ahn, Microporous Mesoporous Mater., 2008, 113, 31–40 CrossRef CAS.
- C. Chen, J. Kim, D. A. Yang and W. S. Ahn, Chem. Eng. J., 2011, 168, 1134–1139 CrossRef CAS.
- P. L. Qin, H. W. Lei, X. L. Zheng, Q. Liu, H. Tao, G. Yang, W. J. Ke, L. B. Xiong, M. C. Qin, X. Z. Zhao and G. J. Fang, Adv. Mater. Interfaces, 2016, 3, 1500799 CrossRef.
- Y. Jeon, D. H. Park, J. I. Park, S. H. Yoon, I. Mochida, J. H. Choy and Y. G. Shul, Sci. Rep., 2013, 3, 2902 CrossRef PubMed.
- Y. P. Chen, K. Ding, L. Yang, B. Xie, F. Q. Song, J. G. Wan, G. G. Wang and M. Hana, Appl. Phys. Lett., 2008, 92, 173112 CrossRef.
- V. Zelenak, M. Badanicova, D. Halamova, J. Cejka, A. Zukal, N. Murafa and G. Goerigk, Chem. Eng. J., 2008, 144, 336–342 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta06969c |
| This journal is © The Royal Society of Chemistry 2017 |