
Design rules for the preparation of low-cost hole transporting materials for perovskite solar cells with moisture barrier properties†
 *a
Johan C.
Bijleveld,
b
Maximilian T.
Sirtl,
a
Yinghong
Hu,
a
Theo J.
Dingemans,
bc
Thomas
Bein
a
and
Pablo
Docampo
*ad
*a
Johan C.
Bijleveld,
b
Maximilian T.
Sirtl,
a
Yinghong
Hu,
a
Theo J.
Dingemans,
bc
Thomas
Bein
a
and
Pablo
Docampo
*ad
Abstract
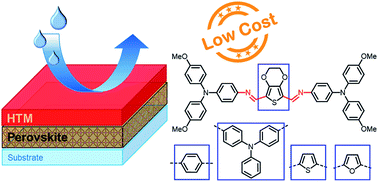
The current state-of-the-art hole transporting materials (HTM) for perovskite solar cells are generally synthesized via cross-coupling reactions that require expensive catalysts, inert reaction conditions and extensive product purification, resulting in high costs and therefore limiting large-scale commercialisation. Here we describe a series of HTMs prepared via simple and clean Schiff-base condensation chemistry with an estimated material cost in the range of 4–54 $ per g. The optoelectronic and thermal properties of the materials are linked to the changes in the chemical structure of the HTMs, which allow us to extract design rules for new materials, supported by density functional theory calculations. Charge transport measurements show hole mobilities in the range of 10−5 to 10−7 cm2 V−1 s−1. Upon addition of LiTFSI the HTMs can be oxidized, resulting in a large increase in the conductivity of the hole transporting layer (HTL). When employed as HTL in perovskite solar cells, power conversion efficiencies close to those of spiro-OMeTAD are obtained. In particular, devices prepared with Diazo-OMeTPA show a higher open-circuit voltage. Furthermore, we show that azomethine-based HTMs can act as effective moisture barriers, resulting in a significant increase in the stability of the underlying perovskite film. We assign the improved properties to the presence of a dipole in our molecules which promotes a close molecular packing and thus leads to a high density of the as-formed HTM films, preventing the ingress of water. This work shows that HTMs prepared via condensation chemistry are not only a low-cost alternative to spiro-OMeTAD, but also act as a functional barrier against moisture-induced degradation in perovskite solar cells.
 Please wait while we load your content...
Please wait while we load your content...

