
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A comparative study of anion-exchange membranes tethered with different hetero-cycloaliphatic quaternary ammonium hydroxides†
Hai-Son
Dang
and
Patric
Jannasch
 *
*
Department of Chemistry, Polymer and Materials Chemistry, Lund University, P.O. Box 124, Lund 221 00, Sweden. E-mail: patric.jannasch@chem.lu.se; Fax: +46-46-222-4012
First published on 2nd October 2017
Abstract
Quaternary ammonium (QA) cations with high alkaline stability are crucial for the long term performance of anion-exchange membrane (AEM) fuel cells. Here, we have tethered poly(phenylene oxide) (PPO) with 8 different hetero-cycloaliphatic QA cations via pentyl spacer chains. The thermal and alkaline stabilities, as well as hydroxide ion conductivity, were systematically evaluated with the primary aim to identify degradation reactions and establish cation design principles. The study included AEMs functionalized with 1-methylazepanium, 1-methylpyrrolidinium, 1-methylmorpholinium, quinuclidinium, as well as 1-methyl-, 1,4-dimethyl-, 1,3,5-trimethyl-, and 1,2,6-trimethylpiperidinium, all within a narrow ion exchange capacity (IEC) range. For reference, PPO was also functionalized with trimethylammonium and dipropylmethylammonium cations on pentyl spacers, and with trimethylammonium and 1-methylpiperidinium QAs in benzylic positions directly on the PPO backbone. The alkaline stability of hetero-cycloaliphatic QA cations was found to depend critically on their position in the polymer structure, ring size, the presence of an additional heteroatom and ring substitution pattern. For example, 1,2,6-trimethylpiperidinium and 1-methylazepanium degraded via Hofmann elimination and 1-methylmorpholinium via ring opening by both Hofmann elimination and substitution reactions, while no degradation was detected by 1H NMR spectroscopy of other cations after 16 days in 1 M NaOH at 90 °C. The hydroxide ion conductivity of the AEMs in the study reached values between 64 and 150 mS cm−1 at 80 °C, depending on the cation and IEC. AEMs tethered with piperidinium and quinuclidinium cations via pentyl spacers were found to show the best overall properties. Hence, the combined results provide insights that may guide the selection of cationic groups and membrane materials to improve the durability and performance of alkaline electrochemical energy conversion and storage devices.
Introduction
Fuel cells are electrochemical devices that offer prospects of zero-emission automobiles, as well as stationary and portable power generation. In this context, the anion-exchange membrane fuel cell (AEMFC) has emerged as a viable alternative to the proton-exchange membrane fuel cell (PEMFC), which has been extensively developed over the last decade. Operating under alkaline conditions with the hydroxide ion as the charge carrier, the AEMFC offers several important advantages such as faster oxygen reduction reaction kinetics at the cathode and the possibility to use non-platinum metal catalysts such as silver, cobalt and nickel.1–6 Nevertheless, the development of the AEMFC is currently severely hampered by the lack of anion-exchange membranes (AEMs) which have the necessary combination of high alkaline stability and sufficient hydroxide ion conductivity to provide long-term high performance of the system.6,7The hydroxide ion is strongly basic and nucleophilic, and is likely to react with the quaternary ammonium (QA) cations that are tethered to the polymer of the AEM. Under strongly alkaline conditions, QA cations are known to degrade via nucleophilic substitution, Hofmann β-elimination and different rearrangement reactions, including Stevens rearrangement in the absence of β-hydrogens.8–10 Hence, the degradation rate not only depends on the nature of the QA cation, but also on the structure of the polymer backbone and the precise positions of the cations in the polymeric structure. For example, electron-withdrawing groups in backbones such as polysulfones activate attack by hydroxide ion which results in chain scissions.11,12 Poly(phenylene oxide) contains only phenylene rings linked by ether bridges and is therefore significantly more stable under alkaline conditions.6 Many early studies on AEMs frequently employed benzyltrimethylammonium cations, which are readily and inexpensively formed by reacting benzylbromide or -chloride groups on the polymer backbones with trimethylamine. However, several reports have shown that these cations are likely to degrade via nucleophilic substitution,9,11 and that adjacent ether linkages are activated for hydrolysis.12,13 In an early seminal work, Tomoi et al. demonstrated that cations placed on polystyrene via flexible alkyl spacer units had a significantly higher alkaline stability than corresponding benzyltrimethylammonium cations.14 This finding has inspired a number of different synthetic approaches to tether QAs to polymer backbones via alkyl spacers.15–30 In general, the results have shown that these AEMs have significantly higher degree of alkaline stability, and an improved level of ionic clustering and ionic conductivity, in comparison with corresponding AEMs with benzyltrimethylammonium cations positioned directly on the backbone.21,22,30 By using density functional theory calculations, Pivovar and coworkers showed that Hofmann elimination is the most likely degradation pathway for alkyl trimethylammonium cations.31,32 Still, because of steric effects, the energy barrier against elimination increased dramatically when the length of the alkyl chain was increased from 2 to 4 methylene groups and stabilized from 4 to 6.32
Numerous different QA cations have been attached to polymer backbones and evaluated in AEMs under alkaline conditions. This includes various tetraalkylammonium, benzimidazolium, imidazolium, pyridinium and guanidinium cations.19,33–38 Studies of model compounds can provide important information about stability and degradation reactions, although it is not obvious that low-molecular weight QA cations have the identical or similar alkaline stability to the same QA cations tethered to a polymer in an AEM. Marino and Kreuer have systematically studied the alkaline stability of a large number of low molecular weight ammonium model compounds, and identified hetero-cycloaliphatic QAs as particularly long-lived.10N,N-Dimethylpiperidinium and N,N-dimethylpyrrolidinium were found to have half-times of 87 and 37 h, respectively, in 6 M NaOH at 160 °C, while the benzyltrimethylammonium cation only reached 4.2 h under the same conditions. The high alkaline stability of the hetero-cycloaliphatic QAs has been explained by a low ring strain and that the conformational restrictions imposed by the ring configuration increases the transition state energy of both substitution and elimination reactions.10 QAs with bicyclic aliphatic spirane structures showed even longer halftimes, up to 110 h for 6-azonia-spiro[5.5]undecane in 6 M NaOH at 160 °C.10 However, the incorporation of these spirocyclic QA cations into polymeric structures represents a significant synthetic challenge, although a few recent examples have been reported by us39–41 and others.42 Polymers and AEMs functionalized with mono-cycloaliphatic QAs are much more accessible and represent a viable alternative to the commercial AEMs of today.
In the present work we have systematically investigated the alkaline stability and performance of AEMs based on PPO functionalized with 8 different hetero-cycloaliphatic QAs via pentyl spacer units. The hetero-cycloaliphatic QAs were selected to study the effect of ring size, methyl substitution pattern and the presence of additional heteroatoms in the ring, and included 1-methylazepanium, 1-methylpyrrolidinium, quinuclidinium, 1-methylmorpholinium, as well as 1-methyl-, 1,4-dimethyl-, 1,3,5-trimethyl-, and 1,2,6-trimethylpiperidinium. PPO polymers with QAs in benzylic positions directly on the backbone were also prepared in order to investigate the effect of the placement in the polymeric structure. Moreover, PPO was functionalized with non-cyclic trimethylammonium and dipropylmethylammonium cations on pentyl spacers for reference. All the polymers of the study had a similar ion exchange capacity which allowed a direct comparison of the performance of the different hetero-cycloaliphatic QAs. AEMs of the different polymers were cast from solution and investigated with respect to morphology (ionic clustering), thermal and alkaline stability, water uptake and hydroxide ion conductivity in order to study and compare overall membrane performance and to identify the most important degradation pathways.
Experimental section
Materials
Tetrahydrofuran (THF, HPLC grade, Honeywell) was dried over molecular sieves (Acros; 4 Å, 8–12 mesh) before use. Poly(2,6-dimethyl-1,4-phenylene oxide) (PPO, Sigma-Aldrich, Mn = 20 kg mol−1, MwMn−1 = 2.3), n-butyllithium (n-BuLi, 2.5 M, solution in hexanes, Acros), dimethyl sulfoxide-d6 (99+%, Sigma-Aldrich), chloroform-d (99+%, Sigma-Aldrich), 1,6-dibromohexane (98%, Acros), N-methyl-2-pyrrolidone (NMP, reagent grade, Acros), trimethyl amine (TMA, 7.3 M aq. solution, Acros), quinuclidine (Qui, 97%, Sigma-Aldrich), N-methylpiperidine (Pip, 99%, Sigma-Aldrich), N-methylpyrrolidine (Pyr, 97%, Sigma-Aldrich), 4-methylmorpholine (Mor, 98+%, Sigma-Aldrich), azepane (Aze, 99%, Sigma-Aldrich), 2,6-dimethylpiperidine (2,6DMPip, 98%, Sigma-Aldrich), 3,5-dimethylpiperidine (3,5DMPip, 96+%, Sigma-Aldrich), 4-methylpiperidine (4MPip, 96+%, Sigma-Aldrich), N-methyldipropylamine (DPMA, 98%, Sigma-Aldrich), iodomethane (99%, Sigma-Aldrich), diethyl ether (99+%, Sigma-Aldrich), sodium nitrate (99%, Sigma), sodium bromide (99.5%, Sigma-Aldrich), methanol (MeOH, HPLC grade, Honeywell) and 2-propanol (IPA, HPLC grade, Honeywell) were all used as received. Silver nitrate (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 995%, Sigma-Aldrich) was dried under vacuum at 30 °C in the dark before use.
995%, Sigma-Aldrich) was dried under vacuum at 30 °C in the dark before use.
Bromoalkylation of PPO
A sample of PPO modified with bromopentyl side chains was prepared via lithiation and subsequent bromoalkylation, as described previously (Scheme 1).22 The degree of bromination (DB) was targeted at ∼30 bromoalkyl chains per 100 repeating units of PPO by controlling the degree of lithiation. A 4-neck 500 mL round bottomed flask equipped with a thermometer and a mechanical stirrer fitted with an argon/vacuum line was filled with PPO (4 g, 33.29 mmol repeat units) and dry THF (400 mL). The content was heated to 60 °C and kept for 40 min to fully dissolve the PPO and was then allowed to cool to room temperature before being degassed using 7 vacuum/argon cycles. A few droplets of n-BuLi solution were slowly added until the solution turned pale yellow in order to consume any remaining reactive impurities. Next, 24 mL of n-BuLi (60 mmol) was added dropwise and the color quickly turned dark-yellow and insoluble lithiated PPO began to precipitate out of the solution. The reaction mixture was then heated to 55 °C and left for 3 h with stirring until a homogeneous orange-red solution formed. After cooling to room temperature, the flask was transferred to an isopropanol/dry ice bath for 30 min to cool to −70 °C. The lithiated PPO was then quenched by quickly injecting a 200% molar excess of 1,4-dibromohexane (based on the amount of added n-BuLi). The bromoalkylation reaction occurred immediately, followed by a change of color back to pale yellow. The solution was left stirring overnight until finally obtaining a colorless solution to ensure complete reaction. Next, the solution was added dropwise to methanol to obtain a fine white precipitate of the product, designated as PPO5-Br. After filtration, the polymer was washed repeatedly with methanol and dried under vacuum at 50 °C until a constant weight was obtained. The organolithium synthesis was straightforward and efficient and the bromoalkylation reaction resulted in a very high yield (∼100%). No gel formation was observed when dissolving the PPO5-Br in common solvents such as THF, NMP and chloroform.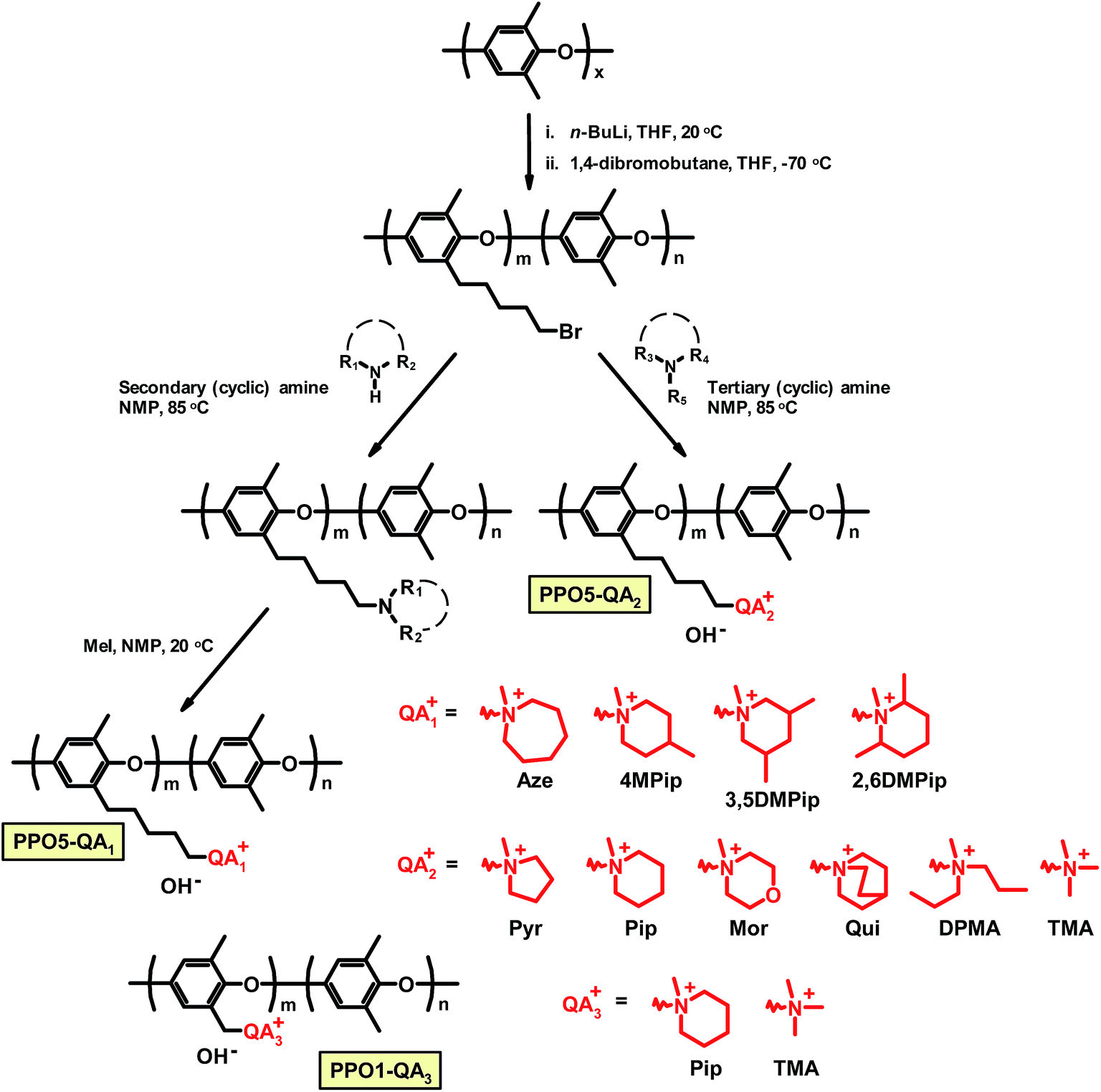 | ||
| Scheme 1 Preparation of PPO functionalized with different QA cations either by reaction of bromoalkylated PPO with the corresponding secondary (cyclic) amine, followed by reaction with iodomethane (samples PPO5-QA1+), or by direct reaction with corresponding tertiary (cyclic) amines (samples PPO5-QA2+). In addition, PPO samples with QA cations in benzylic positions (samples PPO5-QA3+) were prepared by reacting benzyl brominated PPO with the corresponding tertiary amine. All polymers were subsequently ion-exchanged to the OH− form after AEM fabrication. | ||
Introduction of QA cations
Two different pathways were employed to functionalize PPO with 10 different QA cations on pentyl alkyl spacers (Scheme 1). This included 8 different hetero-cycloaliphatic QA cations. Because the same bromoalkylated precursor PPO was used throughout the work, the ion exchange capacity (IEC) varied only slightly, mainly depending on the different molecular weights of the QA cations. Consequently the IEC was kept in a narrow range (1.72–1.90 mequiv. g−1, see Table 1) for all AEMs.The first pathway involved a two-step synthesis in which the Br atoms of PPO5-Br were first displaced by reaction with the corresponding secondary amine, followed by a quaternization reaction with iodomethane. In this way PPO was tethered with 1,4-dimethylpiperidinium (designated as PPO5-4MPip), 1,3,5-trimethylpiperidinium (PPO5-3,5DMPip) and 1,2,6-trimethylpiperidinium (PPO5-2,6DMPip), as well as 1-methylazepanium (PPO5-Aze). This pathway is illustrated by the synthesis of the latter sample in which 0.8 g of PPO5Br-31 (1.38 mmol of Br) and 0.41 g of azepane (4.14 mmol, 200% excess) were dissolved in 16 g of NMP to form a homogeneous solution. After heating to 85 °C, the solution was stirred for 7 days to fully substitute the terminal Br atoms with tertiary azepane rings. The homogeneous solution was then poured into diethyl ether (500 mL) and the intermediate was collected by filtration, washed intensively with diethyl ether, and then dried under vacuum at room temperature overnight. Next, the polymer was weighed and dissolved in NMP to form a 5 wt% solution before adding an excess amount of iodomethane (>200%). The solution was stirred for 2 days in the dark and then added dropwise into diethyl ether to precipitate PPO5-Aze. To convert to the Br− form, the sample was soaked in 200 mL of 1 M aq. NaBr at 50 °C under stirring for at least 48 h. After washing intensively with de-ionized water, the product was dried under vacuum for at least 2 days before further use.
Following the second pathway, the QA cations were introduced by directly substituting the Br atoms of PPO5-Br in Menshutkin reactions with the corresponding tertiary amine. In this way, PPO was tethered with 1-methylpyrrolidinium (designated as PPO5-Pyr), 1-methylpiperidinium (PPO5-Pip), 1-methylmorpholinium (PPO5-Mor), quinuclidinium (PPO5-Qui), dipropylmethylammonium (PPO5-DPMA) and trimethylammonium (PPO5-TMA) QAs, respectively. The latter two samples were included as reference materials to the samples carrying the different hetero-cycloaliphatic QA cations. The preparation of PPO5Q-Qui is described here. An amount of 16 g of NMP was first added to 0.8 g of PPO5-Br (1.38 mmol of Br) and 0.47 g of quinuclidine (4.14 mmol, 200% excess) and the mixture was stirred to form a homogeneous solution. Next, the solution was kept at 85 °C for 7 days under stirring to ensure complete quaternization. The homogeneous orange-red solution was added dropwise into diethyl ether (500 mL) and the product was collected by filtration, washed repeatedly with diethyl ether, and dried under vacuum at room temperature to a constant weight.
In addition, a PPO containing an average of 23 bromobenzyl groups per 100 repeating units (PPO1-Br, DB = 23) was reacted with N-methylpiperidine and trimethylamine, respectively, to introduce 1-methylpiperidinium and trimethylammonium QAs in samples PPO1-Pip and PPO1-TMA, respectively. The preparation followed our previously reported method,21,23 and the DB value of the precursor polymer was chosen to reach IECs close to the other samples in the present study (Table 1).
Structural characterization
1H NMR spectra of the different polymers were recorded in chloroform-d (δ = 7.26 ppm) or DMSO-d6 (δ = 2.50 ppm) solutions, using a Bruker DRX 400 spectrometer operating at 400.13 MHz. The number and weight average molecular weight values (Mn and Mw, respectively) of the brominated PPOs were evaluated using HPLC-grade chloroform as an eluent at room temperature in a size-exclusion chromatography (SEC) system with a series of three Shodex gel columns (KF-805, −804 and −802.5) and a refractive index detector. Standard polystyrenes of known molecular weights with low polydispersities (Mn = 650 kg mol−1 from Water Associates, and Mn = 96 and 30 kg mol−1 from Polymer Laboratories, and Mn = 3.18 kg mol−1 from Agilent Technologies) were used for the calibration. An amount of 5 mg dry polymer was first dissolved in 1 mL of chloroform and passed through a Teflon filter with pore size 0.45 μm. Finally, 200 μL of the sample solution was injected and analyzed at a flow rate of 1.0 mL min−1.Membrane preparation
An amount of 0.15 g of each cationic polymer in the Br− form was dissolved in 3 g of NMP to form a homogeneous solution. After pouring the solution onto a Petri dish, AEM formation was achieved in a well-ventilated casting oven at 80 °C in 48 h. The membrane was removed from the dish, washed repeatedly, and stored in de-ionized water before further measurements. In order to obtain the OH− form, the AEM was immersed in 1 M aq. NaOH and kept for at least 48 h under a nitrogen atmosphere. The NaOH solution was exchanged at least 3 times to ensure complete removal of any traces of remaining Br− ions. The AEMs had an average thickness of ∼70 μm.Thermal characterization
The thermal stability of the polymers was studied using a TA Instruments TGA Q500 under a nitrogen atmosphere. The sample was first dried in a vacuum oven at 50 °C for at least 48 h. Next, the sample was kept isothermally at 150 °C for 10 min in the TGA to remove any traces of absorbed water. TGA measurements were performed at a heating rate of 10 °C min−1 in the temperature interval 50–600 °C under a 60 mL min−1 nitrogen flow. The thermal decomposition temperature (Td,95) was noted at 5% weight loss. DSC measurements were performed with a model Q2000 DSC analyzer from TA Instruments to determine the glass transition temperature (Tg) of the brominated precursor polymers. Data were collected during the following cycle: 20 → 280 → 50 → 280 °C. The upper limit of 280 °C was based on the decomposition temperature obtained by TGA.Determination of ion exchange capacity and water uptake
The theoretical IEC (IECNMR) values of the AEMs in the Br− form were calculated from the 1H NMR spectrum of the PPO5-Br and PPO1-Br precursors, assuming that the respective quaternization reaction reached full conversion. The IECNMR values in the OH− form were also calculated assuming that all Br− ions of the polymer were exchanged to OH− ions. The experimental IEC (IECtitr) of the AEMs was determined by Mohr titrations as follows. The sample was dried under vacuum for at least 48 h. Next, 0.050 g of the sample was stored in 0.2 M aq. NaNO3 (25.00 mL) under stirring for at least 48 h until all Br− ions were completely exchanged from the sample. A 5.0 mL volume of the solution was taken and titrated with 0.01 M aq. AgNO3 using K2CrO4 as the indicator. The end point of the titration was observed by the formation of the Ag2CrO4 precipitate, followed by a color change from yellow to stable reddish brown. The procedure was repeated 3 times to calculate the average value of IECtitr.The dry weight of the AEM sample in the Br− form (WBr) was measured after drying at 50 °C for at least 48 h in a vacuum oven. Based on the value of the IECtitr and the dry weight WBr, the dry weight in the OH− form (WOH) was calculated. The wet weight in the OH− form was measured by the following procedure. The dried sample in the Br− form was first kept in a large excess of degassed 1 M aq. NaOH solution for at least 24 h under a nitrogen atmosphere. The NaOH solution was exchanged twice during this period. Next, the sample was immersed in 200 mL de-ionized water for at least 2 h under a nitrogen atmosphere. The water used was repeatedly exchanged (4–6 times) with fresh de-ionized water every 2 h until the rinsed water reached neutral pH. The sample was then kept in de-ionized water for an additional 24 h under a nitrogen flow. Finally, the wet weight in the OH− form (W′OH) was noted after wiping off excess water with tissue paper. The water uptake (WUOH) was calculated as:
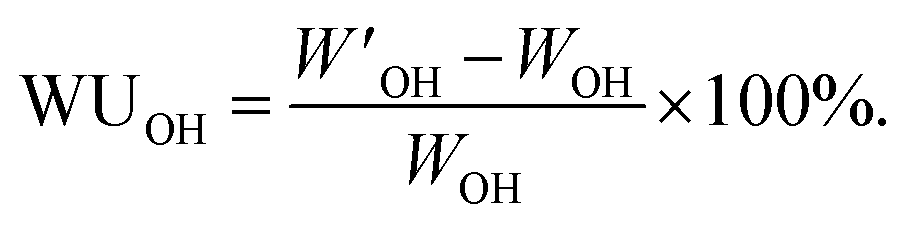 | (1) |
Subsequently, the membrane in the OH− form was recovered by immersing the sample in 1 M degassed aq. NaOH solution again for at least 4 h. Using a similar procedure to that described above, the water uptake was measured at 40, 60, and 80 °C, respectively by soaking the sample in de-ionized water under argon at different temperatures.
The hydration number (λ), i.e. the number of water molecules per QA cation, was calculated as:
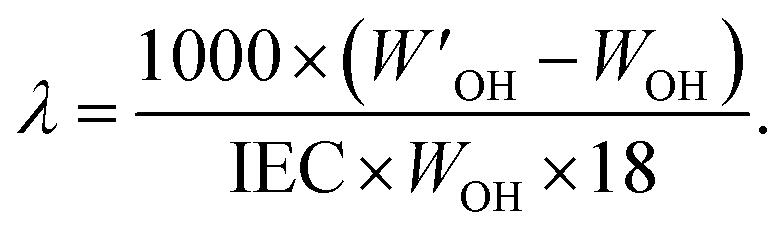 | (2) |
Conductivity measurements
The OH− conductivity (σOH) measurements were performed by electrochemical impedance spectroscopy (EIS) employing a Novocontrol high resolution dielectric analyzer 1.01S in the frequency range 10−1 to 10−7 Hz at 50 mV. The membrane sample was first cut into a 1.4 cm × 1.4 cm piece and soaked in a closed vial containing 100 mL 1 M aq. NaOH for 48 h under nitrogen to obtain the AEM in the OH− form. The NaOH solution was exchanged with fresh solutions three times. Subsequently, the sample was transferred into a beaker containing 200 mL of de-ionized water and kept for at least 2 h under a nitrogen atmosphere. The rinsing water was repeatedly exchanged (4–6 times) with new fresh de-ionized water every 2 h until it reached neutral pH. Finally, OH− conductivity measurements were performed under immersed conditions using a two-probe method during the cycle: 20 → 80 → −20 → 80 °C.Small angle X-ray scattering
The morphology (ionic clustering) of the AEMs was analyzed by small-angle X-ray scattering (SAXS) measurements. First, the membrane in the Br− form was cut into a 2 cm × 0.5 cm piece and dried under vacuum at 50 °C for 24 h. Next, SAXS measurements were carried out using a SAXSLab ApS instrument (JJ-Xray, Denmark) equipped with a microfocus sealed X-ray tube and a Pilatus detector. The scattering vector (q) was calculated as: | (3) |
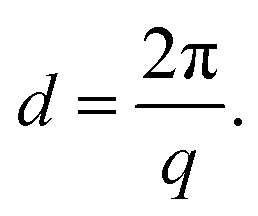 | (4) |
Evaluation of alkaline stability
The alkaline stability was studied by monitoring the structural degradation of the membrane polymers by 1H NMR spectroscopy. Three sample pieces of in total ∼10 mg were cut from each membrane and soaked in 350 mL of degassed 1 M aq. NaOH under a nitrogen atmosphere at 90 °C. The NaOH solution was exchanged with fresh solution every 3 days. After specified time periods of 4, 8 and 16 days, respectively, a sample piece was taken out and washed extensively with de-ionized water to remove any residual NaOH. In order to convert to the Br− form, the sample was soaked in 1 M aq. NaBr at 60 °C for at least 48 h under stirring. The NaBr solution was exchanged with fresh solutions at least 3 times during the ion exchange process. Next, any residual NaBr salt in the sample was removed by extensive washing and storing in de-ionized water under stirring for 24 h. Prior to the NMR measurements, the sample was first dried for at least 24 h at 50 °C under vacuum, before gently dissolving it in DMSO-d6 under heating. To complement the NMR data, the alkaline stability was also studied by TGA analysis. Three weighed samples (∼10 mg) were treated with an excess of degassed 1 M aq. NaOH under nitrogen at 90 °C for 4, 8 and 16 days, respectively. The next steps followed the same procedure described above for the NMR evaluation, including the ion-exchange and drying. However, the final AEM sample (∼3 mg) was directly analyzed by TGA, as described above.Results and discussion
Polymer synthesis and characterization
PPO was tethered with QA cations by displacing the bromine atoms of PPO functionalized with bromopentyl side chains with the respective amine (Scheme 1). The brominated precursor was synthesized by lithiation of native PPO in THF using n-BuLi, followed by reaction with an excess of 1,4-dibromobutane, as previously reported.22 In this procedure, PPO is lithiated in both aromatic and benzylic positions. However, only the latter positions are reactive towards dibromoalkanes under the conditions used, thus resulting in the formation of the bromopentyl side chains.22,43–45 The expected chemical structure of the product (PPO5-Br) was confirmed by 1H NMR analysis, and the degree of bromination (DB, the average number of brominated sites per 100 PPO repeating units) was calculated to be 31 by comparing the integrated signals arising from the aromatic protons at 6.4 ppm and from the –CH2Br protons at 3.4 ppm (Fig. 1). As seen in Table 1, the polydispersity of PPO5-Br increased noticeably in relation to the neat PPO because of limited branching reactions during the bromoalkylation, most likely because of di-substitution of a fraction of the dibromobutane added.22 Still, no gel formation was detected. By using the same PPO5-Br as the precursor for all the polymers tethered with QA cations on pentyl spacers, and achieving close to quantitative displacement, a series of samples was produced with different QAs but with the same attachment of the cations and with IEC values within a narrow range (1.72–1.90 mequiv. g−1). In order to study the effect of the attachment of the QAs, a precursor PPO was functionalized with benzylbromide groups (PPO1-Br, DB = 23). This sample was prepared by radical-mediated benzylic bromination of PPO using NBS and AIBN, as described previously.21 The Tg of PPO1-Br was found to increase by 16 °C in relation to the neat PPO, presumably the result of increased polymer chain stiffness after introduction of the benzylbromide groups (Table 1). In contrast, the Tg of PPO5-Br was 67 °C below that of the neat PPO because of internal plasticization induced by the flexible bromopentyl side chains.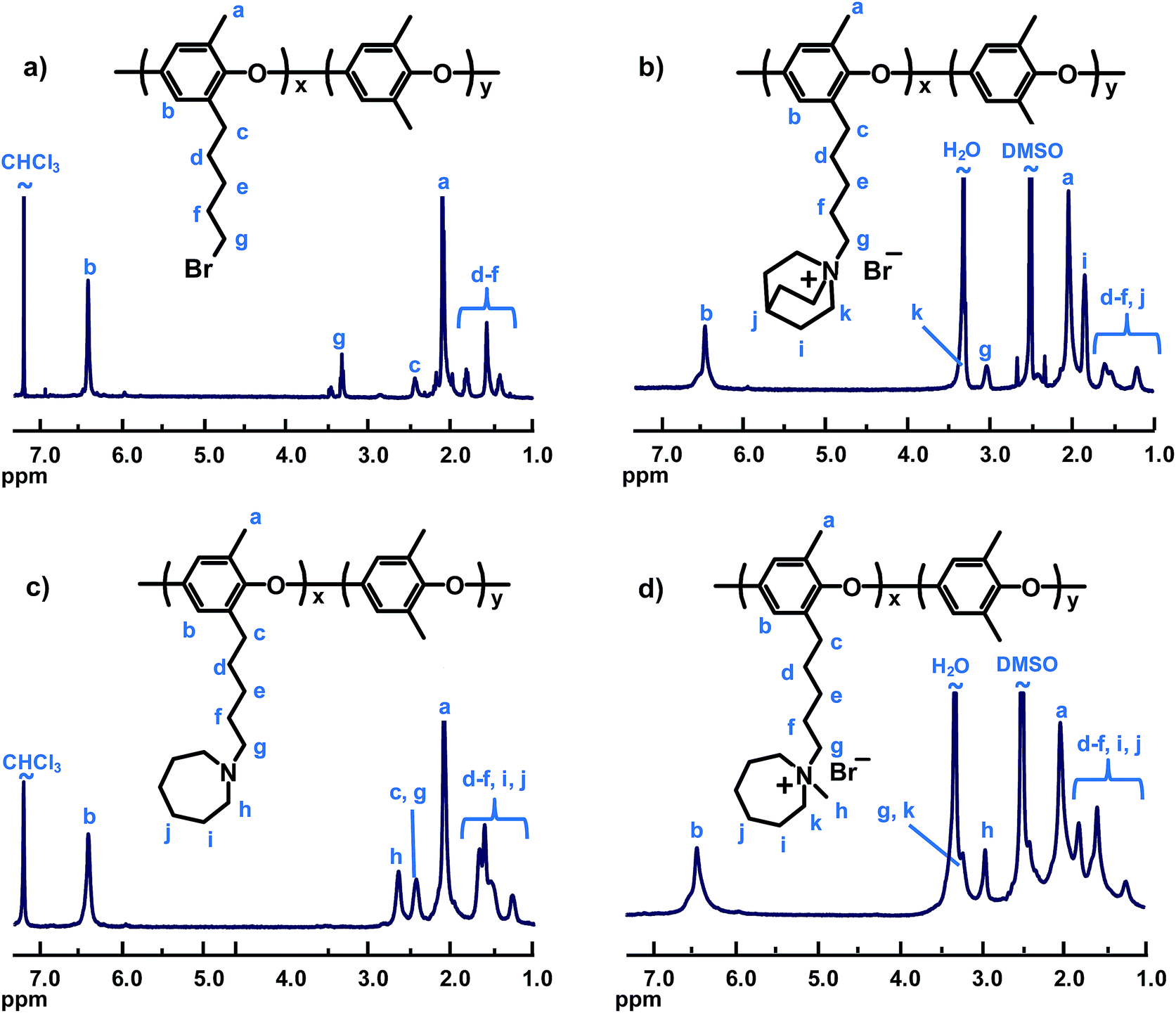 | ||
| Fig. 1 Representative 1H NMR spectra of modified PPO samples: (a) bromoalkylated PPO5-Br, (b) PPO5-Qui, (c) intermediate to PPO5-Aze before quaternization, and (d) PPO5-Aze. | ||
Starting from PPO5-Br and PPO1-Br, PPO was functionalized with the different cations according to Scheme 1, either in a direct Menshutkin reaction involving the corresponding tertiary amine (N-methylpyrrolidine, N-methylpiperidine, N-methylmorpholine, quinuclidine, N-methyldipropylamine and trimethylamine, respectively), or via reaction with the corresponding secondary amine (azepane, 4-methylpiperidine, 3,5-dimethylpiperidine and 2,6-dimethylpiperidine, respectively), followed by a Menshutkin reaction with iodomethane. These reactions proceeded efficiently in NMP and resulted in virtually complete displacement of bromine atoms and introduction of the various QA cations, as confirmed by Mohr titrations and 1H NMR analysis. Representative 1H NMR spectra of intermediates and products of the two quaternization routes are shown in Fig. 1 (additional spectra available as ESI, Fig. S1–S4†). As expected, the spectra of the PPOs with cyclic QAs on heptyl spacers shared many common features. Signals of the aromatic and benzylic methyl protons of the PPO appeared at approximately 6.4 and 2.2 ppm, respectively. The α-protons of the cationic centers gave rise to signals at ∼2.9 ppm (–CH3), 3.2 ppm (ring and spacer –CH2–, overlapped by the H2O signal) and ∼3.6 ppm (ring –CH–, PPO5-3,5DMPip). Additional ring and spacer –CH2– signals were observed in between 1.2 and 2.5 ppm. The many signals in this region resulted in extensive signal overlapping. Still, the integration and comparison of the aromatic and non-overlapped –CH2– ring signals indicated complete conversion within the error of the method. The samples with QAs in benzylic positions showed distinct signals at ∼4.25 ppm from the benzylic protons (Fig. S3†).
Membrane morphology
Approximately 70 μm thick transparent and yellowish AEMs were cast from NMP solutions of the different cationic polymers at 80 °C over 2 days. After drying, the morphology of the AEMs in the Br− form was studied by small angle X-ray scattering (SAXS). Phase separation and formation of distinct ionic clusters during membrane formation seems to be essential in order to form percolating water-rich domains for efficient ion and water transport.46 The presence of ionic clusters can be studied by SAXS by analyzing so-called ionomer peaks. Calculated from the position of the ionomer peak (qmax), the characteristic separation distance (d) has been correlated with the average distances between ion-rich domains in AEMs.46 As seen in Fig. 2, the positions of the ionomer peaks of all the AEMs fall within a narrow range, around qmax = 2.1–2.2 nm−1. This corresponded to d = 3.0–2.8 nm (Table 2). Hence, the qmax value was essentially independent of the nature and placement of the QA. Based on the scattering intensity, i.e., the height of the ionomer peak, the level of organization was lowest in PPO1-Pip, and in PPO5-Aze, PPO5-DPMA and PPO5-2,6DMP, indicating that the degree of ionic clustering was low when the QAs were placed in benzylic positions and when the cation was comparatively large. This is in agreement with previous observations.22 The SAXS profiles of PPO5-Pyr, -Pip and -Qui hinted a higher degree of ionic clustering than the previously mentioned group of AEMs, and were very similar to that of PPO5-TMA. Notably, the scattering of PPO5-Pip was more intense than that of PPO1-Pip. Finally, the scattering intensity of PPO5-Mor implied the highest degree of ionic clustering. Perhaps the presence of an additional oxygen atom in the ring increased the polarity, and hence the propensity to form ionic clusters.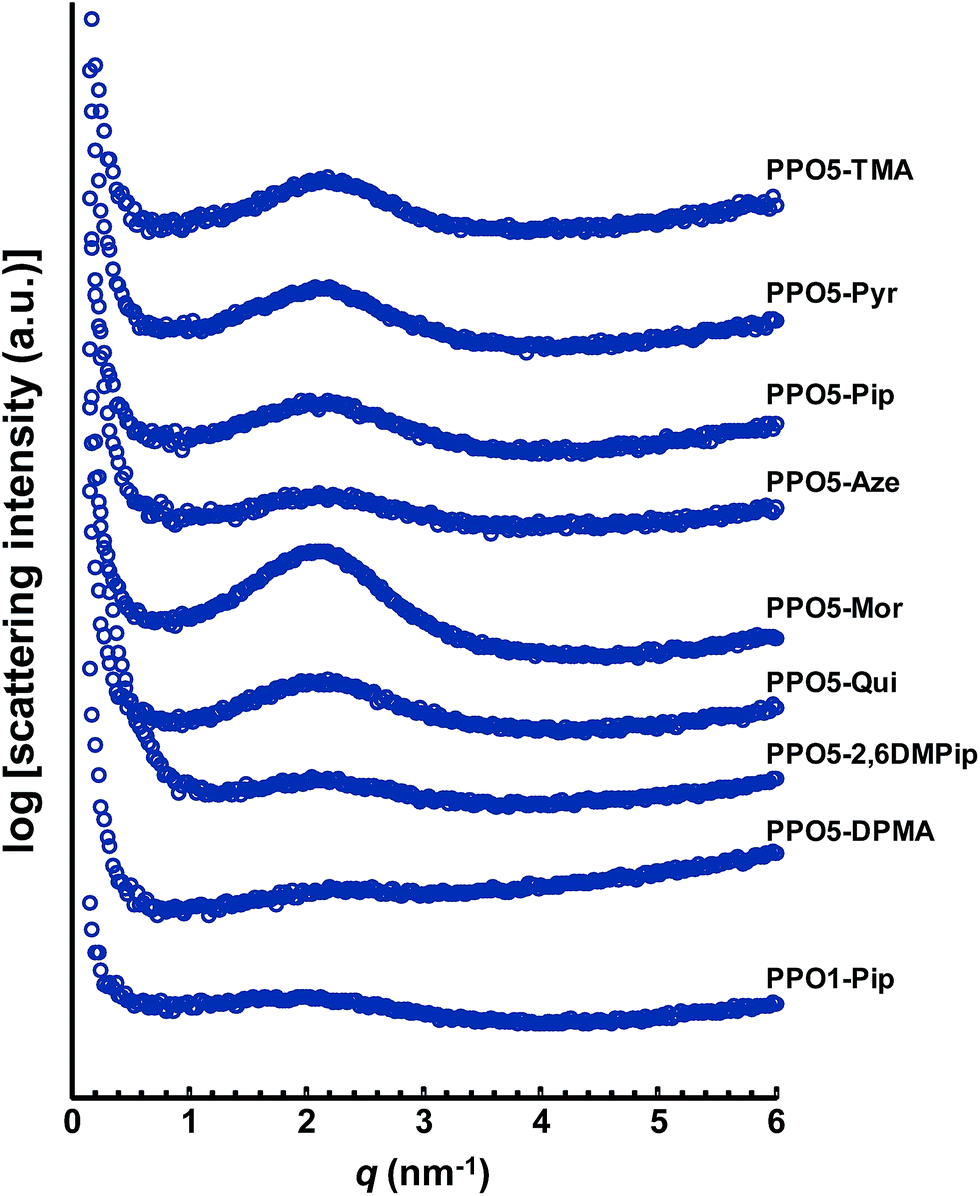 | ||
| Fig. 2 SAXS profiles of dry AEMs based on PPOs functionalized with different hetero-cycloaliphatic QAs in the Br− form. For clarity, the curves have been shifted vertically (qmax and d values collected in Table 2). | ||
| AEM | Precursor polymer | IECNMRa [mequiv. g−1] | IECtitra [mequiv. g−1] | T d,95 [°C] | q max [nm−1] | d [nm] | λ | σ OH (80 °C) [mS cm−1] | Alkaline degradationf |
|---|---|---|---|---|---|---|---|---|---|
| a IEC in the OH− form (values within parenthesis are for the Br− form). b Measured by TGA under N2. c Measured by SAXS on dry AEMs. d Immersed in the OH− form at 20 °C. e OH− conductivity, AEMs fully hydrated. f After 384 h in 1 M NaOH at 90 °C (by 1H NMR analysis). n.m. = not measured. n.d. not detected. | |||||||||
| PPO5-Pip | PPO5-Br | 1.84 (1.65) | 1.78 (1.60) | 264 | 2.1 | 3.0 | 17 | 117 | n.d. |
| PPO5-Pyr | PPO5-Br | 1.84 (1.65) | 1.78 (1.60) | 248 | 2.1 | 2.9 | 14 | 89 | n.d. |
| PPO5-Aze | PPO5-Br | 1.78 (1.60) | 1.72 (1.55) | 231 | 2.2 | 2.8 | 15 | 78 | Hofmann elimination |
| PPO5-Mor | PPO5-Br | 1.84 (1.65) | 1.78 (1.60) | 227 | 2.1 | 3.0 | 14 | 101 | Hofmann elimination/nucleophilic substitution |
| PPO5-Qui | PPO5-Br | 1.72 (1.55) | 1.78 (1.60) | 283 | 2.1 | 3.0 | 13 | 151 | n.d. |
| PPO5-4MPip | PPO5-Br | 1.78 (1.60) | 1.72 (1.55) | 249 | n.m. | n.m. | 16 | 94 | n.d. |
| PPO5-3,5DMPip | PPO5-Br | 1.72 (1.55) | 1.66 (1.50) | 236 | n.m. | n.m. | 17 | 52 | n.d. |
| PPO5-2,6DMPip | PPO5-Br | 1.72 (1.55) | 1.66 (1.50) | 231 | 2.0 | 3.2 | 15 | 86 | Hofmann elimination |
| PPO5-TMA | PPO5-Br | 1.90 (1.70) | 1.90 (1.70) | 253 | 2.1 | 3.0 | 15 | 139 | n.d. |
| PPO5-DPMA | PPO5-Br | 1.78 (1.60) | 1.72 (1.55) | 210 | 2.2 | 2.9 | 10 | 64 | n.d. |
| PPO1-TMA | PPO1-Br | 1.67 (1.51) | 1.60 (1.45) | 247 | 2.1 | 3.0 | 9 | 14 | Nucleophilic substitution |
| PPO1-Pip | PPO1-Br | 1.57 (1.43) | 1.48 (1.35) | 276 | 2.0 | 3.2 | 9 | 11 | Nucleophilic substitution |
Water uptake
The water in hydrated AEMs facilitates ionic dissociation (i.e. formation of charge carriers) and ion transport and thus has a profound effect on the measured ionic conductivity. Still, excessive water uptake will result in loss of mechanical properties and dilution of the charge carriers, resulting in reduced conductivity. Fig. 3 shows the water uptake of the AEMs in the OH− form under fully hydrated (immersed) conditions between 20 and 80 °C. In the present case, the water uptake can be expected to depend on the size of the QA group, i.e., a large QA group resulting in a comparatively lower IEC value, and the ionic distribution in the AEM, i.e. the level of ionic clustering. In the studied IEC range (1.7–1.9 mequiv. g−1) a small change in the IEC value typically has a large effect on the water uptake.23 As expected, the water uptake increased with temperature. Between 20 and 40 °C, the water uptake increased moderately, while in the range 60–80 °C the increase with temperature was higher. In general, AEMs with a combination of efficient ionic clustering, i.e. showing distinct ionomer peaks, and comparatively high IEC tended to reach high water uptake values. Examples included PPO5-Pip, -Pyr, and -TMA. The only striking exception to this trend was PPO5-Mor, which reached just above 50 wt% at 80 °C, despite a relatively high IEC and pronounced ionomer peak. The AEMs functionalized with methylated piperidinium cations, i.e., PPO5-4MPip, -3,5DMPip and -2,6DMPip, took up less water than the non-methylated PPO5-Pip, presumably because of the more hydrophobic character of the former. With their poor ionic clustering and low IEC values, the AEMs with QA cations in benzylic positions only reached low water uptake values.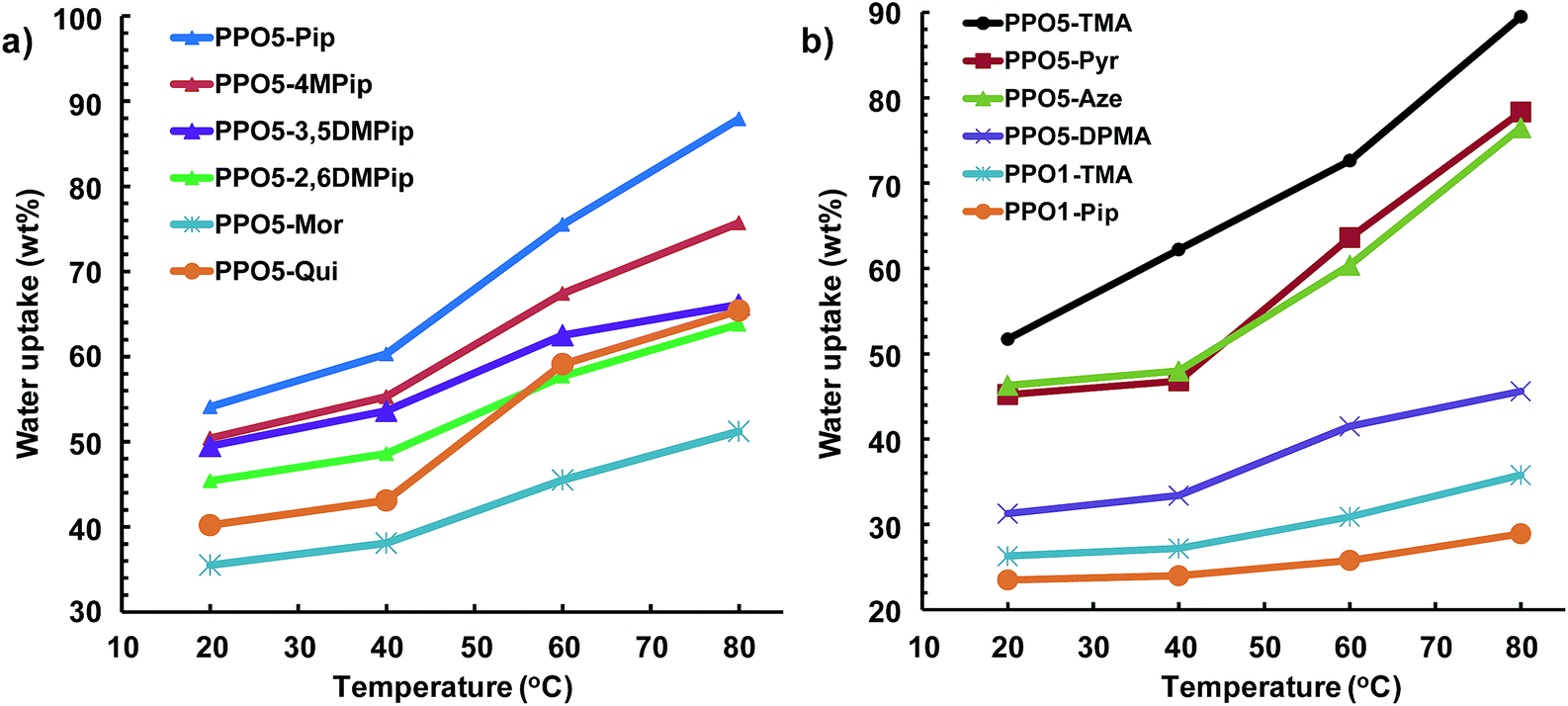 | ||
| Fig. 3 Water uptake of fully hydrated (immersed) AEMs in the OH− form as a function of temperature (data shown in two plots for clarity, note the different scales on y-axes). | ||
OH− conductivity
The ionic conductivity of AEMs depends on a complex interplay between ionic concentration (IEC) and water content, as well as on morphological factors (such as the degree of percolation) and the degree of ionic dissociation.46–48 In the present case, the ionic conductivity of fully hydrated AEMs in the OH− form was measured by EIS between −20 and 80 °C using a two-probe cell. The formation of carbonates via uptake of CO2 was avoided by carefully washing and equilibrating the AEMs in degassed distilled water under a N2 atmosphere. As expected, the OH− conductivity data showed an increase with temperature (Fig. 4). The conductivity increased especially between −20 and 0 because of the melting of ice in the AEMs.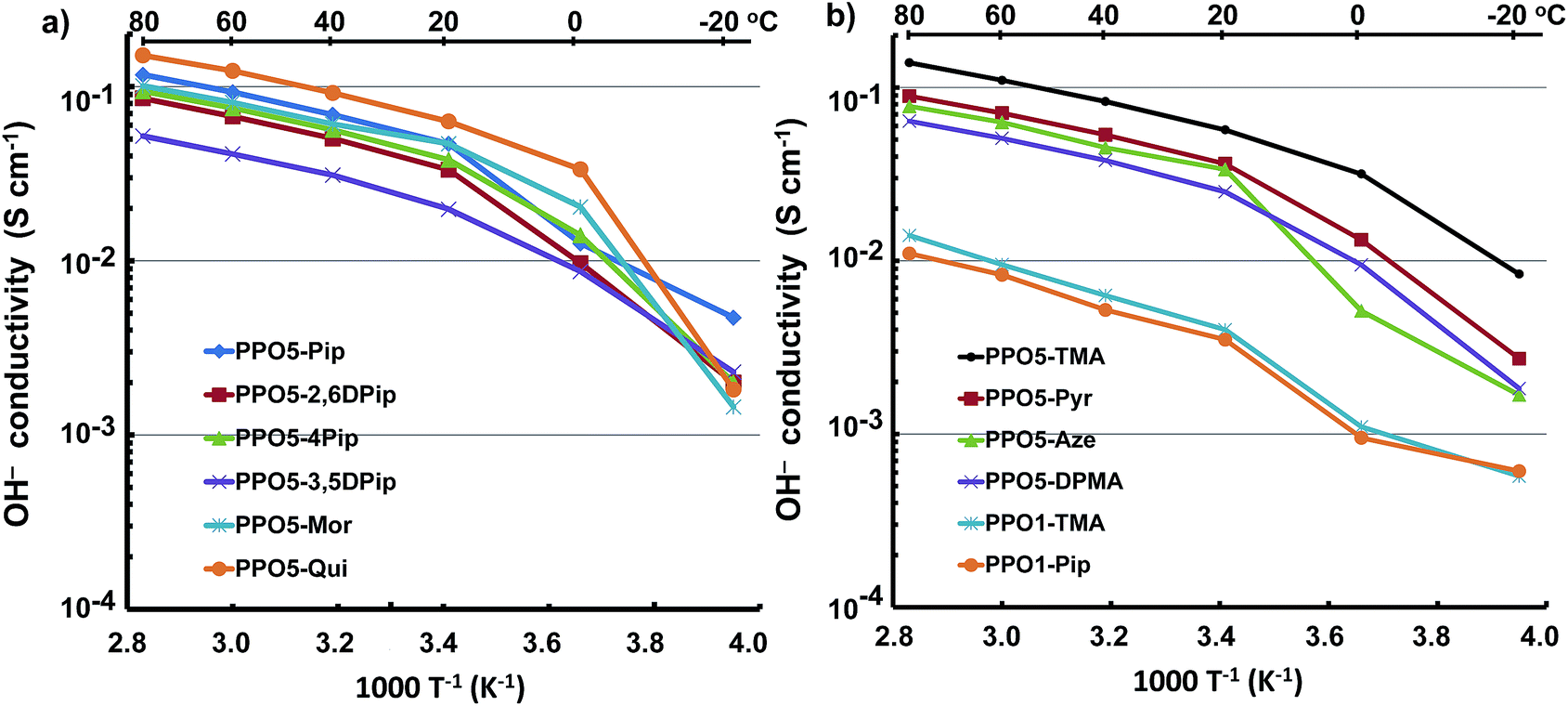 | ||
| Fig. 4 Arrhenius plots of the OH− conductivity of AEMs as a function of temperature. Data measured by EIS with the AEMs fully hydrated (immersed) and shown in two plots for clarity. | ||
In general, the AEMs displayed high levels of conductivity, between 54 and 151 mS cm−1 at 80 °C. In particular, PPO5-Qui, -TMA, -Mor and -Pip reached above 100 mS cm−1, and PPO5-Pyr, -Aze, -Mor, -4MPip, -2,6DMPip, and -DPMA showed conductivities above 60 mS cm−1 at 80 °C. Membrane PPO5-3,5DMPip displayed the lowest conductivity at 54 mS cm−1 at the same temperature. At the same IEC value, PPO5-Aze had a higher conductivity (78 mS cm−1 at 80 °C) than PPO5-DPMA (64 mS cm−1), indicating a higher conductivity for the AEM with hetero-cycloaliphatic cations. This may be explained by the poor ability of PPO5-DPMA to form ionic clusters, in combination with its significantly lower water uptake. Comparing the AEMs based on hetero-cycloaliphatic cations with different ring sizes, the conductivity increased in the order: PPO5-Aze < PPO5-Pyr < PPO5-Pip. The high value of the latter AEM may be explained by a combination of efficient ionic clustering and high water uptake. The conductivity of PPO5-Pip was also higher than that of PPO5-Mor, which showed efficient ionic clustering but low water uptake. The oxygen atom of morpholinium draws away electrons inductively across the entire σ bonded ring, and hence the degree of ion dissociation can be expected to be lower than for the piperidinium cation. This may explain the comparatively low water uptake and conductivity of this AEM. Substituting the piperidinium ring with methyl groups decreased the IEC and water uptake, and this was reflected in the conductivity which decreased in the order: PPO5-Pip > PPO5-4MPip > PPO5-2,6DMPip > PPO5-3,5DMPip.
PPO1-TMA and -Pip showed low and very similar conductivities, presumably because of their low IEC values, poor ionic clustering and low water uptake. Thus, the conductivities of these two AEMs reached only 14 and 11 mS cm−1 at 80 °C. The poor performance of AEMs with QAs in benzylic positions, in comparison to those with QAs on alkyl spacers, was in agreement with previous observations.15–30
Thermal stability
The thermal decomposition of the AEMs in the Br− form was studied by TGA under a nitrogen atmosphere after a drying period at 150 °C to remove any residual water. The thermal decomposition temperature (Td,95) was recorded at 5% weight loss and the values of the different AEMs are collected in Table 2 (all TGA traces shown in Fig. S7 and S8†). The decomposition of all the materials occurred in two steps, with the first resulting from the decomposition of the different QA cations and the second one from the degradation of the PPO backbone.21–24 While the second step was noted at approximately 400 °C for all the AEMs, the Td,95 values (connected to the first decomposition step) varied characteristically with the nature of the QA cation.For the QAs with different ring sizes, Td,95 increased by 33 °C in the order PPO5-Aze < PPO5-Pyr < PPO5-Pip, indicating the importance of low ring strain for the thermal stability. The presence of an additional oxygen atom in the hetero-cycloaliphatic QAs proved detrimental for the stability as PPO5-Mor decomposed 37 °C below PPO5-Pip. The stability of the piperidinium cation clearly decreased with the degree of methyl substitution with PPO5-Pip at Td,95 = 264 °C and PPO5-4MPip, -3,5DMPip and -2,6DMPip at Td,95 = 249, 236 and 231 °C, respectively. Quinuclidinium may also be regarded as a substituted piperidinium, and PPO5-Qui showed the highest thermal stability in the series with Td,95 = 283 °C. Presumably, the cage-like and symmetrical quinuclidinium cation significantly contributed to the excellent thermal stability. The influence of the placement of the QAs on the thermal stability was inconclusive from the results. While Td,95 of PPO1-Pip was 12 °C above the value of PPO5-Pip, the value of PPO1-TMA was 6 °C below that of PPO5-TMA.
Comparing the hetero-cycloaliphatic QAs with the non-cyclic ones, both PPO5-Pip and PPO5-Qui decomposed at significantly higher temperatures than PPO5-TMA. Moreover, PPO1-Pip decomposed 29 °C above PPO1-TMA. Attaching butyl chains to the cationic center decreased the stability considerably and PPO5-DPMA displayed the lowest Td,95 value in the series at 210 °C. The combined results demonstrated that AEMs based on hetero-cycloaliphatic QAs may reach significantly higher thermal stability than corresponding AEMs with standard non-cyclic ones. However, the cyclic QAs may be destabilized by ring strain, substitutions and the presence of additional heteroatoms.
Alkaline stability
Achieving sufficient long-term alkaline stability is today the most critical challenge when it comes to the development of AEMs for alkaline membrane fuel cells and electrolyzers.6,25 We have previously reported that no degradation was detected for PPO5-TMA after immersion in 1 M NaOH at 80 °C for 8 days.22 In the present study, to increase the severity of the conditions, the alkaline stability of the different AEMs was investigated after immersion in 1 M aq. NaOH at 90 °C after 4, 8 and 16 days. After cooling to room temperature, the samples were removed from the NaOH solution, washed with water and ion-exchanged to the Br− form. After drying, the samples were dissolved in DMSO-d6 for 1H NMR analysis. The alkaline stability was then evaluated by comparing NMR spectra (both signal positions and intensities) before and after the immersion periods. In addition, new signals were interpreted to identify the mode of QA degradation. Signal overlapping complicated the analysis, especially in the region of the methylene proton signals (1–2.5 ppm) and around the DMSO and water signals.
Fig. 5 shows the 1H NMR spectra of all the PPOs tethered with hetero-cycloaliphatic QAs before and after storage of the respective AEM in the alkaline solution for 16 days (384 h). As seen, some of the AEMs were found to degrade under these conditions, confirmed by new signals emerging from degradation products, while no degradation was detected for other AEMs. Regarding AEMs functionalized with QAs with different ring sizes, no degradation was detected for PPO5-Pip and -Pyr, while PPO5-Aze was clearly found to degrade via Hofmann ring-opening elimination. As seen in the spectrum of the latter after the alkaline treatment (Fig. 5c), two new signals emerged at 4.9 ppm (2 protons) and 5.8 ppm (1 proton) and were attributed to the formation of –N(CH3)–(CH2)4–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2. Apparently, the 7-membered ring does not possess the rigidity necessary to hinder Hofmann degradation. While no degradation was detected for PPO5-Pip, PPO5-Mor showed obvious signs of degradation (Fig. 5d). The morpholinium cation was observed to degrade via both Hofmann ring-opening elimination and nucleophilic ring-opening substitution reactions. After 384 h immersion, the signal intensities of this QA cation appeared to decrease. In parallel, two new signals started to appear at 4.1 and 3.6 ppm, respectively. The signal at 4.1 ppm (two protons) was assigned to the formation of –N(CH3)–CH2–CH2–O–CHCH2 groups as a result of the Hofmann elimination, and the new signal at 3.6 ppm (three protons) most probably arose from the formation of –N(CH3)–CH2–CH2–O–CH2–CH2–OH groups as a result of nucleophilic ring-opening substitution.
CH2. Apparently, the 7-membered ring does not possess the rigidity necessary to hinder Hofmann degradation. While no degradation was detected for PPO5-Pip, PPO5-Mor showed obvious signs of degradation (Fig. 5d). The morpholinium cation was observed to degrade via both Hofmann ring-opening elimination and nucleophilic ring-opening substitution reactions. After 384 h immersion, the signal intensities of this QA cation appeared to decrease. In parallel, two new signals started to appear at 4.1 and 3.6 ppm, respectively. The signal at 4.1 ppm (two protons) was assigned to the formation of –N(CH3)–CH2–CH2–O–CHCH2 groups as a result of the Hofmann elimination, and the new signal at 3.6 ppm (three protons) most probably arose from the formation of –N(CH3)–CH2–CH2–O–CH2–CH2–OH groups as a result of nucleophilic ring-opening substitution.
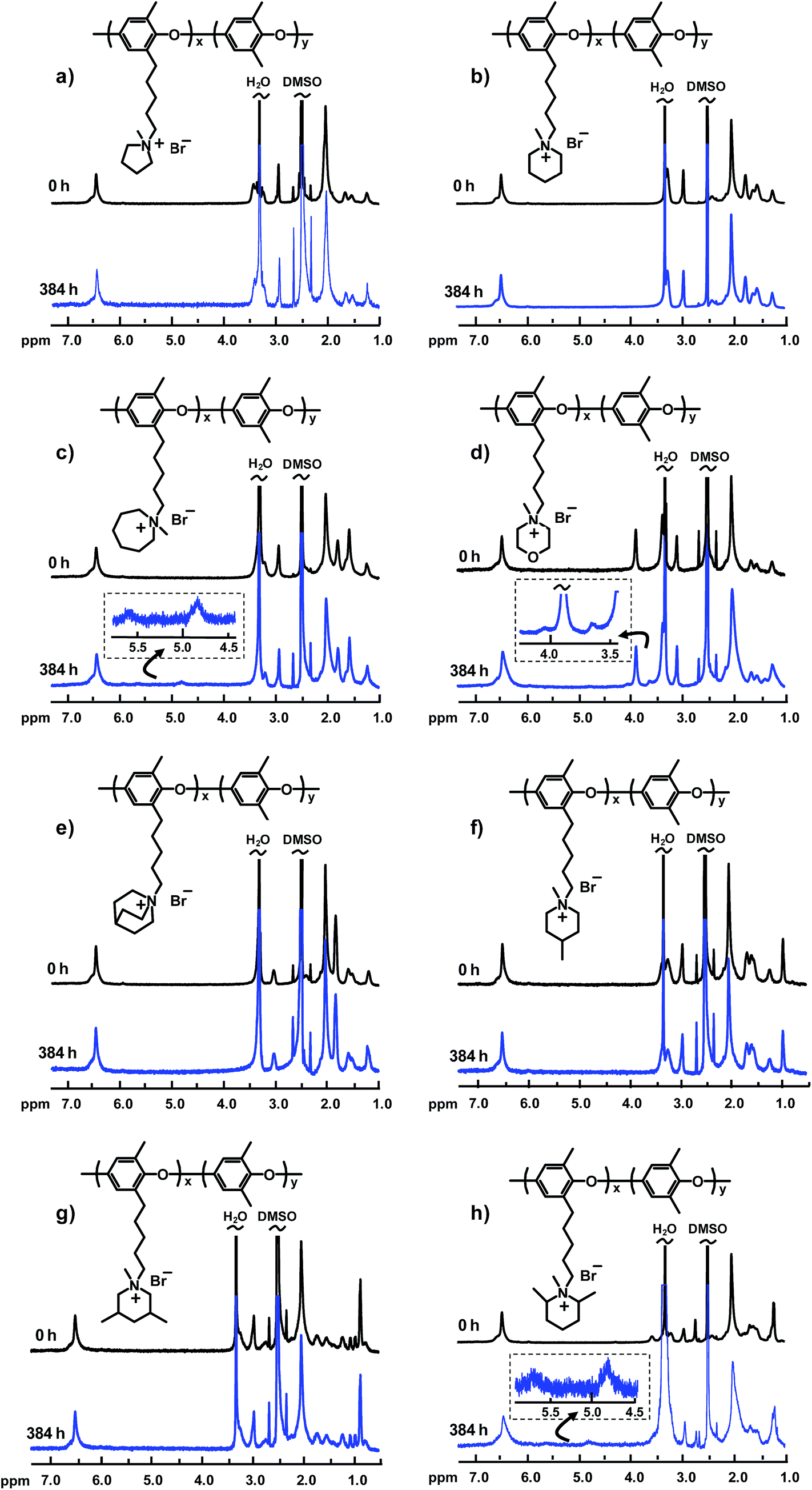 | ||
| Fig. 5 1H NMR spectra of PPOs tethered with different hetero-cycloaliphatic QAs via alkyl spacer chains, before and after immersion of the respective AEM in 1 M aq. NaOH at 90 °C for 384 h: (a) PPO5-Pyr, (b) PPO5-Pip, (c) PPO5-Aze, (d) PPO5-Mor, (e) PPO5-Qui, (f) PPO5-4MPip, (g) PPO5-3,5DMPip, and (h) PPO5-2,6DMPip. Spectra were recorded in DMSO-d6 after ion-exchange to the Br− form. | ||
The results on the AEMs carrying methyl-substituted piperidinium cations revealed no traces of degradation for PPO5-4MPip and -3,5DMPip, whereas new signals emerged in the spectrum of PPO5-2,6DMPip (Fig. 5f–h). As seen in Fig. 5h, the two new signals visible at 4.9 ppm (2 protons) and 5.8 ppm (1 proton) most probably resulted from Hofmann ring-opening elimination and the formation of –N(CH3)–(CH)(CH3)–(CH2)3–CHCH2 units. Thus, it was obvious that the 6 additional (non-ring) β-protons added by the methyl groups in the 2- and 6-positions significantly destabilized the piperidinium ring by enabling efficient Hofmann elimination.
The results on the AEMs with the different substituted piperidinium rings also hinted which positions in the ring are suitable to covalently attach this QA to polymers and AEMs without compromising alkaline stability. Notably, the corresponding spectra of the AEMs functionalized with non-cyclic QAs via pentyl spacers, i.e., PPO5-TMA and -DPMA, revealed no signs of degradation after storage in the alkaline solution (Fig. S10†). This demonstrated that the hetero-cycloaliphatic cation has to be carefully selected in order to acquire advantages over non-cyclic ones.
Fig. 6 shows the spectra from the alkaline stability evaluation of the AEMs with QAs in benzylic positions (PPO1-TMA and -Pip). As seen, the signal intensity of the respective QA cation quickly decreased with immersion time and the polymers lost their solubility in DMSO due to the extensive loss of ionic groups. Hence, the 1H NMR analysis of these materials was performed only after 96 and 192 h, respectively. Even so, the solubility was limited which resulted in low signal-to-noise ratios. As reported previously, QAs attached to benzylic positions are usually prone to fast degradation due to the benzylic attack by OH−, while QAs attached via flexible alkyl spacers are significantly more stable.21,22
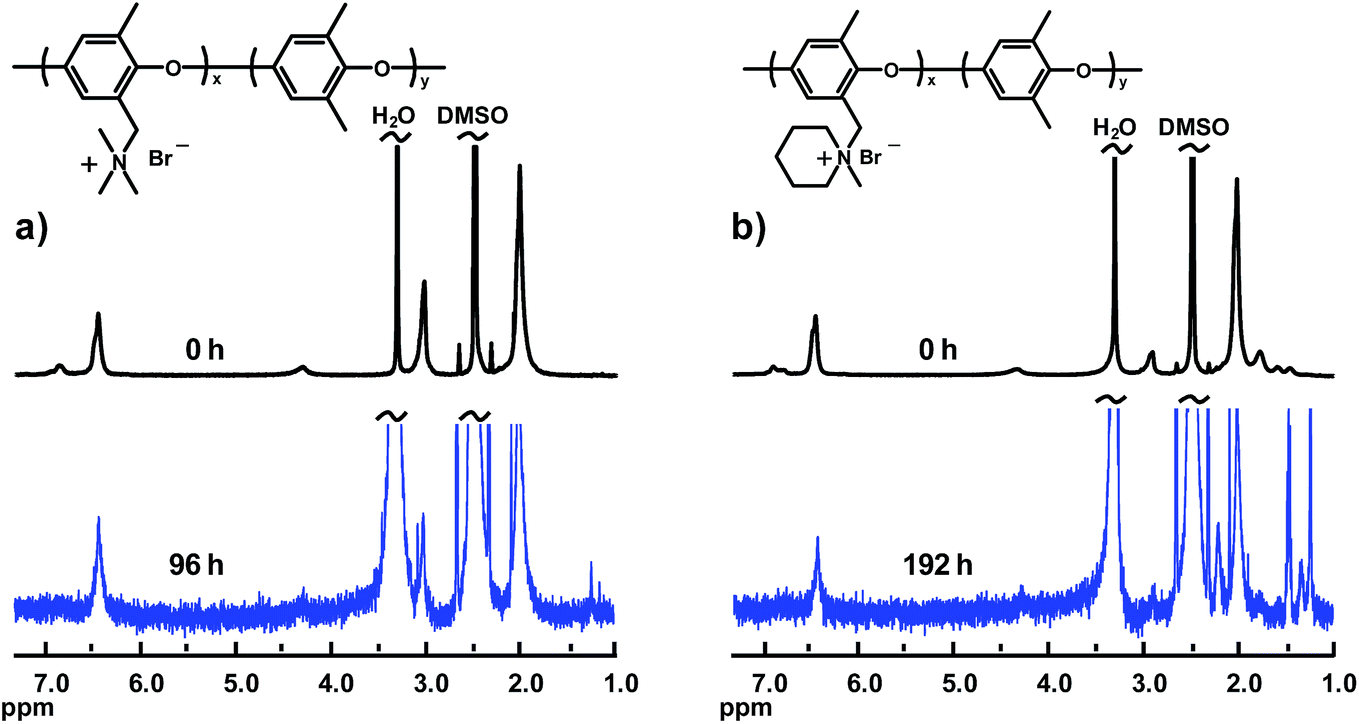 | ||
| Fig. 6 1H NMR spectra of PPOs with QAs in benzylic positions before and after immersion of the respective AEM in 1 M aq. NaOH at 90 °C for 96 and 192 h, respectively: (a) PPO1-TMA and (b) PPO1-Pip. Spectra were recorded in DMSO-d6 after ion-exchange to the Br− form. | ||
To complement the 1H NMR data, the AEMs before and after testing were also analyzed by TGA measurements, which are sensitive to the QA content. Fig. 7 displays the TGA traces of three AEMs where no degradation was detected by the NMR analysis (Fig. 7a–c) and three AEMs where the NMR spectra showed degradation and loss of the respective QA group (Fig. 7d–f). As seen, the thermograms of PPO5-TMA, -Pyr and -Pip show close to identical traces before and after testing (within the error of the method), although it is not possible to exclude small losses of QA cations in any of the samples. In contrast, the traces of PPO5-Aze, -2,6DMPip and PPO5-Mor indicated a significant decrease of the first degradation step, connected with the decomposition of the QA functional side chains, after storage in 1 M aq. NaOH at 90 °C over 384 h. Thus, the TGA results were in very good agreement with the findings by NMR spectroscopy and supported the conclusions of this study.
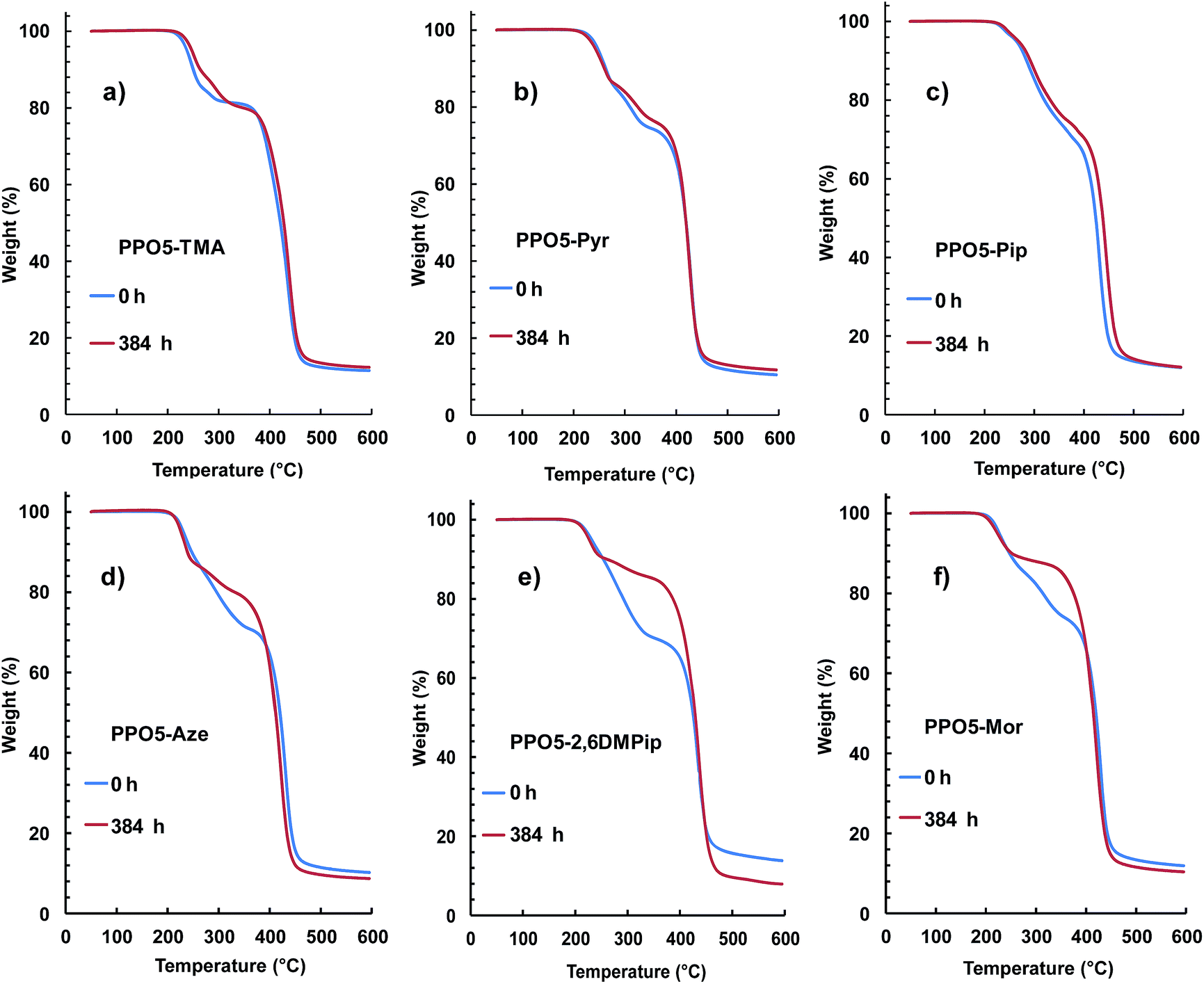 | ||
| Fig. 7 Representative TGA traces of AEMs before and after immersion in 1 M aq. NaOH at 90 °C for 384 h: (a) PPO5-TMA, (b) PPO5-Pyr, (c) PPO5-Pip, (d) PPO5-Aze, (e) PPO5-2,6DMPip, and (f) PPO5-Mor. Samples were ion-exchanged to the Br− form and dried before analysis at 10 °C min−1 under N2. | ||
Conclusions
PPO was tethered with 8 different hetero-cycloaliphatic QA cations via pentyl spacer chains to study the effect of ring size, presence of additional heteroatom and methyl substitution pattern on the stability and performance of AEMs within a narrow IEC range. The results showed that the thermal stability decreased with increasing ring strain and methyl substitution, as well as the presence of an electron withdrawing O-heteroatom (morpholinium). Moreover, the alkaline stability was found to decrease with increased (7-membered) ring size, substitution in the 2-and 6-positions in the piperidinium ring, as well as the presence of the additional O-heteroatom. QA cations attached in benzylic positions on the PPO backbone showed the lowest stability in the study, and the spacer concept provided superior stability and conductivity. The quaternary piperidinium and quinuclidinium functionalized materials showed the best overall properties when comparing the different AEMs tethered with hetero-cycloaliphatic QAs on the basis of thermal and alkaline stability, and OH− conductivity. The quinuclidinium-based AEM displayed efficient ion cluster formation and reached the highest conductivity of the studied AEMs at a moderate water uptake, as well as the highest thermal stability. Moreover, this AEM showed no degradation in the alkaline stability test. Apparently, the bridged bicyclic configuration of this cation brings many advantages in the context of AEMs. The drawbacks of this QA cation are the toxicity and high price of the quinuclidine precursor. Thus, the quaternary piperidinium cation may still possess the most attractive combination of properties. Notably, AEMs tethered with the non-cyclic trimethylammonium cation performed very well with high conductivity and stability. The conditions used in the evaluation of the alkaline stability were not severe enough to cause degradation and show the difference between, e.g., the pyrrolidinium, piperidinium and trimethylammonium cations. In summary, the combined results provide a guide for the selection of QA groups and membrane materials to improve the lifetime and performance of alkaline electrochemical energy devices.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the Swedish Energy Agency and the Swedish Research Council for financial support. We are also grateful to Peter Holmqvist for assistance with SAXS measurements and data treatment.Notes and references
- J. R. Varcoe, R. C. T. Slade, G. L. Wright and Y. Chen, J. Phys. Chem. B, 2006, 110, 21041–21049 CrossRef CAS PubMed.
- J. R. Varcoe, R. C. T. Slade, E. L. H. Yee, S. D. Poynton, D. J. Driscoll and D. C. Apperlev, Chem. Mater., 2007, 19, 2686–2693 CrossRef CAS.
- S. Lu, J. Pan, A. Huang, L. Zhuang and J. Lu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20611–20614 CrossRef CAS.
- J. Pan, C. Chen, L. Zhuang and J. Lu, Acc. Chem. Res., 2012, 45, 473–481 CrossRef CAS PubMed.
- X. Wu and K. Scott, J. Power Sources, 2012, 206, 14–19 CrossRef CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. W. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 CAS.
- M. A. Hickner, A. M. Herring and E. B. Coughlin, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1727–1735 CrossRef CAS.
- F. N. Jones and C. R. Hauser, J. Org. Chem., 1962, 27, 1542–1547 CrossRef CAS.
- A. D. Mohanty and C. Bae, J. Mater. Chem. A, 2014, 2, 17314–17320 CAS.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS PubMed.
- C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2490–2495 CrossRef CAS PubMed.
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y.-K. Choe and C. Bae, Macromolecules, 2016, 49, 3361–3372 CrossRef CAS.
- Y.-K. Choe, C. Fujimoto, K.-S. Lee, L. T. Dalton, K. Ayers, N. J. Henson and Y. S. Kim, Chem. Mater., 2014, 26, 5675–5682 CrossRef CAS.
- M. Tomoi, K. Yamaguchi, R. Ando, Y. Kantake, Y. Aosaki and H. Kubota, J. Appl. Polym. Sci., 1997, 64, 1161–1167 CrossRef CAS.
- W. H. Lee, A. D. Mohanty and C. Bae, ACS Macro Lett., 2015, 4, 453–457 CrossRef CAS.
- W. H. Lee, Y. S. Kim and C. Bae, ACS Macro Lett., 2015, 4, 814–818 CrossRef CAS.
- A. D. Mohanty, C. Y. Ryu, Y. S. Kim and C. Bae, Macromolecules, 2015, 48, 7085–7095 CrossRef CAS.
- M. Zhang, J. L. Liu, Y. G. Wang, L. A. An, M. D. Guiver and N. W. Li, J. Mater. Chem. A, 2015, 3, 12284–12296 CAS.
- M. R. Hibbs, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1736–1742 CrossRef CAS.
- J. Wang, H. B. Wei, S. Z. Yang, H. G. Fang, P. Xu and Y. S. Ding, RSC Adv., 2015, 5, 93415–93422 RSC.
- H.-S. Dang, E. A. Weiber and P. Jannasch, J. Mater. Chem. A, 2015, 3, 5280–5284 CAS.
- H.-S. Dang and P. Jannasch, Macromolecules, 2015, 48, 5742–5751 CrossRef CAS.
- H.-S. Dang and P. Jannasch, J. Mater. Chem. A, 2016, 4, 11924–11938 CAS.
- H.-S. Dang and P. Jannasch, J. Mater. Chem. A, 2016, 4, 17138–17153 CAS.
- E. A. Weiber and P. Jannasch, Macromol. Chem. Phys., 2016, 217, 1108–1118 CrossRef.
- Z. J. Yang, J. H. Zhou, S. W. Wang, J. Q. Hou, L. Wu and T. W. Xu, J. Mater. Chem. A, 2015, 3, 15015–15019 CAS.
- B. C. Lin, L. H. Qiu, B. Qiu, Y. Peng and F. Yan, Macromolecules, 2011, 44, 9642–9649 CrossRef CAS.
- Y. Z. Zhuo, A. L. Lai, Q. G. Zhang, A. M. Zhu, M. L. Ye and Q. L. Liu, J. Mater. Chem. A, 2015, 3, 18105–18114 CAS.
- L. Zhu, J. Pan, C. M. Christensen, B. Lin and M. A. Hickner, Macromolecules, 2016, 49, 3300–3309 CrossRef CAS.
- J. Pan, J. Han, L. Zhu and M. A. Hickner, Chem. Mater., 2017, 29, 5321–5330 CrossRef CAS.
- S. Chempath, J. M. Boncella, L. R. Pratt, N. Henson and B. S. Pivovar, J. Phys. Chem. C, 2010, 114, 11977–11983 CAS.
- H. Long, K. Kim and B. S. Pivovar, J. Phys. Chem. C, 2012, 116, 9419–9426 CAS.
- X. Lin, X. Liang, S. D. Poynton, J. R. Varcoe, A. L. Ong, J. Ran, Y. Li, Q. Li and T. Xu, J. Membr. Sci., 2013, 443, 193–200 CrossRef CAS.
- O. I. Deavin, S. Murphy, A. L. Ong, S. D. Poynton, R. Zeng, H. Herman and J. R. Varcoe, Energy Environ. Sci., 2012, 5, 8584–8597 CAS.
- E. A. Weiber and P. Jannasch, J. Membr. Sci., 2015, 481, 164–171 CrossRef CAS.
- J. Fang, Y. Wu, Y. Zhang, M. Lyu and J. Zhao, Int. J. Hydrogen Energy, 2015, 40, 12392–12399 CrossRef CAS.
- X. Lin, L. Wu, Y. Liu, A. L. Ong, S. D. Poynton, J. R. Varcoe and T. Xu, J. Power Sources, 2012, 217, 373–380 CrossRef CAS.
- D. S. Kim, A. Labouriau, M. D. Guiver and Y. S. Kim, Chem. Mater., 2011, 23, 3795–3797 CrossRef CAS.
- T. H. Pham and P. Jannasch, ACS Macro Lett., 2015, 4, 1370–1375 CrossRef CAS.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Am. Chem. Soc., 2017, 139, 2888–2891 CrossRef CAS PubMed.
- J. S. Olsson, T. H. Pham and P. Jannasch, Macromolecules, 2017, 50, 2784–2793 CrossRef CAS.
- L. Gu, H. Dong, Z. Sun, Y. Li and F. Yan, RSC Adv., 2016, 6, 94387–94398 RSC.
- M. Ingratta, M. Elomaa and P. Jannasch, Polym. Chem., 2010, 1, 739–746 RSC.
- A. J. Chalk and T. J. Hoogeboom, J. Polym. Sci., Part A-1: Polym. Chem., 1969, 7, 1359–1369 CrossRef CAS.
- A. J. Chalk and A. S. Hay, J. Polym. Sci., Part A-1: Polym. Chem., 1969, 7, 691–705 CrossRef CAS.
- K. D. Kreuer, J. Membr. Sci., 2001, 185, 29–39 CrossRef CAS.
- J. Pan, C. Chen, Y. Li, L. Wang, L. S. Tan, G. Li, X. Tang, L. Xiao, J. Lu and L. Zhuang, Energy Environ. Sci., 2014, 7, 354–360 CAS.
- J. Pan, Y. Li, J. J. Han, G. W. Li, L. S. Tan, C. Chen, J. T. Lu and L. Zhuang, Energy Environ. Sci., 2013, 6, 2912–2915 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional NMR spectra, TGA traces and DSC data. See DOI: 10.1039/c7ta06029g |
| This journal is © The Royal Society of Chemistry 2017 |